


Epilepsia
RESUMEN: PUNTOS CLAVE PARA EL PEDIATRA DE ATENCIÓN PRIMARIA
SUMMARY: KEY POINTS FOR PRIMARY CARE PEDIATRICIANS
RESUMEN
La epilepsia es una patología frecuente y que genera alarma en las familias. Lo más importante para el diagnóstico es una buena historia clínica para descartar los episodios paroxísticos no epilépticos y encuadrar las crisis en un tipo concreto según su semiología. El electrocardiograma es útil para clasificar el tipo de epilepsia. La neuroimagen, preferiblemente la resonancia magnética, debe realizarse siempre, salvo en las epilepsias idiopáticas. Las crisis epilépticas no precisan tratamiento para ser yuguladas, salvo que duren más de cinco minutos. El tratamiento crónico de la epilepsia debe ser individualizado.
IMPORTANCIA DEL PROBLEMA EN NUESTRO MEDIO
La importancia de la epilepsia en los niños radica en su frecuencia y, sobre todo, en la alarma que provoca en los adultos tanto el diagnóstico como la sospecha de la misma, ya que la mayoría de los cuadros que son diagnóstico diferencial con ella (síncopes, espasmos del sollozo, etc.) también suelen generar mucha angustia en las familias (tabla 1).
Tabla 1. Mostrar/ocultar

La incidencia anual de la epilepsia en menores de 15 años oscila entre 50 y 100 casos/100 000, con una incidencia acumulada a los 20 años del 1% de la población, siendo máxima en el primer año de vida, para ir disminuyendo hasta la adolescencia, etapa en la que vuelve a aumentar. La prevalencia es de 5/10001-3.
Las definiciones clásicas de crisis epiléptica (CE) y epilepsia han sido reevaluadas recientemente4. En la actualidad, una CE es un acontecimiento transitorio de signos y/o síntomas debidos a una actividad neuronal cerebral anormal excesiva o síncrona (no todo lo episódico es epiléptico, ni tan siquiera todo lo convulsivo lo es, volvemos a recordar los síncopes y los espasmos del sollozo). La epilepsia se define como al menos una CE (no necesariamente no provocada) junto a una alteración duradera en el cerebro que aumenta la probabilidad de futuras CE. Con esta definición, no son necesarias dos crisis para el diagnóstico de epilepsia, siempre y cuando exista algo duradero en el cerebro que suponga un riesgo de recurrencia, pero no sería una epilepsia una crisis única con un cerebro funcional y estructuralmente normal. Está definición supone un cambio conceptual muy importante con respecto a la previa (al menos dos CE no provocadas separadas por 24 horas) y está siendo criticada por los epidemiólogos5.
También las clasificaciones clásicas de CE6 y las epilepsias7 han sido revisadas, estableciendo cinco ejes de clasificación8, como puede verse en la tabla 2.
Tabla 2. Mostrar/ocultar

La clasificación etiológica es de gran importancia tanto para el tratamiento como para el pronóstico. Podemos establecer cuatro grandes grupos que nos ayudarán en la toma de decisiones: epilepsias idiopáticas (de presumible base genética, sin lesión anatómica ni neuropatológica asociada; por ejemplo, epilepsia ausencia infantil), epilepsias sintomáticas (epilepsias adquiridas o genéticamente determinadas, con lesión anatómica grosera o patológica clara; por ejemplo, epilepsias secundarias a una malformación cerebral o a una cromosomopatía), epilepsias provocadas (debidas a una causa sistémica concreta o a un factor desencadenante; por ejemplo, las crisis febriles o las fotoinducidas) y epilepsias criptogénicas (se les supone una causa, pero esta se desconoce)9.
MANEJO DIAGNÓSTICO
En el diagnóstico de la epilepsia lo fundamental es la anamnesis. Solo el relato preciso del episodio y las circunstancias en las que ocurrió diferencian una CE de otros episodios (tabla 1). La posible exposición a tóxicos y los antecedentes personales y familiares también deben recogerse de forma precisa. Debemos realizar una exploración general y neurológica meticulosas. La primera es imprescindible para descartar otros procesos. La segunda nos ayudará en la clasificación de la CE entre focal (si encontramos focalidad en la exploración) y generalizada. Ningún signo clínico (por ejemplo, las convulsiones) diferencia al 100% una CE de una no epiléptica5.
La necesidad de realizar una analítica con glucemia, sodio y creatinina después de una primera crisis no febril en niños con recuperación completa está discutida en la literatura10; para algunos no es necesaria, salvo que existan dudas por la historia clínica de que se trate de otro proceso como una hipoglucemia o una hiponatremia5,11. Para otros son necesarios estos datos y también el calcio y el magnesio12.
Por otra parte, si ante un primer episodio afebril dudamos si es o no comicial, una elevación de leucocitos y de creatincinasa en sangre apoyará el diagnóstico de comicialidad11.
Los tóxicos en orina u otras analíticas solo estarán indicadas cuando la historia clínica nos haga sospechar otra patología de base u otros procesos que puedan simular una epilepsia10. En cuanto a la punción lumbar, está claro que no está indicada salvo que se sospeche una infección del sistema nervioso central11.
En resumen, si la historia clínica es muy clara no necesitaremos estudios analíticos, pero con frecuencia no es tan clara y nos obliga a realizarlos. No hay que hacer pruebas diagnósticas a un niño previamente sano con exploración normal que ha sufrido su primera crisis febril, pero tendremos que hacer “todo” a uno comatoso con fiebre alta que ha convulsionado y muy posiblemente dos horas después será un niño con su primera crisis febril.
Una vez que se sospecha una epilepsia, el paciente debe ser derivado lo antes posible a atención especializada13.
El electroencefalograma (EEG) ayuda a determinar el tipo de crisis y de síndrome epiléptico, así como el riesgo de recurrencia después de una primera CE afebril11, pero no es útil para el diagnóstico diferencial entre epilepsia y no epilepsia. Por un lado, hay pacientes epilépticos con EEG normal y hasta un 7% de los niños (del 0,6 al 7%, según los estudios) tiene descargas epileptiformes en el EEG sin ser epilépticos5,14; por tanto, debe evitarse en otros cuadros como los síncopes13, ya que puede ser un factor de confusión y alarma. Un EEG basal debe realizarse lo antes posible. Si con este no llegásemos a clasificar la epilepsia, un EEG de sueño estaría indicado. El vídeo-EEG también estaría en una segunda línea si después de uno convencional seguimos teniendo dudas13.
Todas las guías coinciden en la utilidad de los estudios de neuroimagen en el diagnóstico de la epilepsia, salvo en las epilepsias generalizadas idiopáticas y en las epilepsias focales idiopáticas con paroxismos rolándico-silvianos de buena evolución. También coinciden en la superioridad de la resonancia magnética (RM) sobre la tomografía axial computerizada (TAC)5,10,11,13,15, quedando este último reservado únicamente para situaciones urgentes en las que no se pueda realizar la RM, cuando exista una contraindicación formal para la realización de la RM5,13,15 o en niños a los que haya que sedar para la RM y no para el TAC13, en cuyo caso se valorará la relación riesgo/beneficio, estando indicada la RM siempre en menores de dos años13,15.
La prueba de neuroimagen debe realizarse lo antes posible13. Debe realizarse de forma urgente cuando aparece un déficit neurológico focal que no se resuelve de forma rápida11, hay deterioro del nivel de conciencia, signos de hipertensión intracraneal, signos meníngeos, antecedente de traumatismo craneoencefálico, historia previa de cáncer o inmunodeficiencia. No estaría indicada en un niño con fiebre y exploración neurológica normal o en niños con alteración tóxico-metabólica clara5,10.
MANEJO TERAPÉUTICO
Tratamiento de la crisis epiléptica y del estatus epiléptico
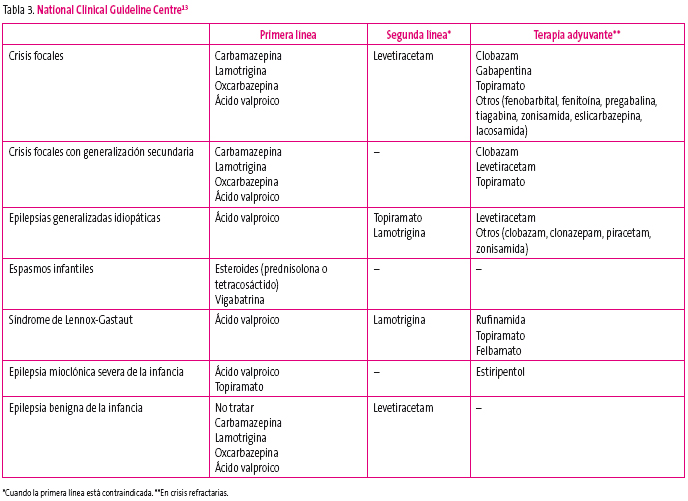
Una CE aislada no precisa tratamiento, ya que suele ser muy breve, pero cuando dura más de 5-10 minutos la posibilidad de remisión espontánea resulta escasa5. Sin embargo, el estatus epiléptico (EE), definido como crisis de más de 30 minutos de duración o crisis repetidas sin recuperación de la conciencia entre ellas, es una emergencia vital16 (tabla 3).
Tabla 3. Mostrar/ocultar
El tratamiento de una CE en la fase aguda puede plantearse en las infecciones del sistema nervioso central, en traumatismos craneoencefálicos graves o si existe una lesión estructural potencialmente epileptógena5.
El tratamiento fundamental de una CE sintomática aguda o provocada (hipoglucemia, hiponatremia, encefalitis, etc.) es el de la etiología que la provocó. El tratamiento sintomático de las crisis debe mantenerse solo durante la fase aguda, días o, en pocas ocasiones, semanas10.
El tratamiento del EE con benzodiazepinas debe realizarse tanto en el ámbito hospitalario como en el extrahospitalario17. En base a la dificultad para remitir espontáneamente las crisis prolongadas se preconiza manejar las CE de más de cinco minutos y a los pacientes con tres o más crisis de igual forma, como emergencias13.
Idealmente por vía intravenosa (IV), ya que esta nos permite estudiar posibles alteraciones analíticas y estabilizar hemodinámicamente al paciente si fuese preciso. El lorazepam (0,1 mg/kg IV) es más eficaz que la combinación de diazepam (0,2 mg/kg IV) y fenitoína (18 mg/Kg IV)18, pero desafortunadamente continúa sin comercializarse en nuestro país. Cuando resulta imposible disponer de vía IV, solemos utilizar diazepam rectal (0,5 mg/kg), pero tenemos otras opciones. El midazolam intramuscular (excelente absorción, no como el diazepam rectal, que la tiene errática) o nasal (0,2 mg/kg) es igual de eficaz que el diazepam IV, y el midazolam oral (0,5 mg/kg) ha demostrado ser más eficaz que el diazepam rectal19, siendo actualmente la opción recomendada13.
Es a estos niños con crisis muy prolongadas a los que prescribiremos diazepam rectal. Aunque en las guías se recomienda también el midazolam oral (explicándole a los padres que está fuera de indicación), en España es muy complicado, al tratarse de un fármaco de uso hospitalario13.
Tratamiento de la epilepsia
El tratamiento del proceso subyacente, si existe, debe realizarse simpre que sea posible10.
Lo primero que debemos recordar es que los fármacos antiepilépticos (FAE) no previenen el desarrollo de la epilepsia ni la curan5,20. Por este motivo, deben utilizarse cuando el beneficio de reducir el número de CE sea superior al riesgo de sus efectos adversos20. El tratamiento de la epilepsia siempre debe ser individualizado5.
El riesgo de recurrencia de una primera CE sin tratamiento es del 40-60% a los dos años en estudios en niños. Si la CE es sintomática (tumor, lesión porencefálica, parálisis cerebral infantil, etc.) el riesgo aumenta, también si el EEG es anormal, y el riesgo es mayor si se dan ambas cosas. Hasta un 50% de los pacientes tratados con FAE puede sufrir efectos adversos y al menos un 10% tiene que suspender la medicación por este motivo5.
Como regla general, trataremos después de dos o más crisis, pero no debemos tratar la primera CE (más de una en 24 horas a efectos de tratamiento crónico con FAE debe considerarse como una CE única), salvo que el riesgo de una posible recurrencia resulte intolerable para la familia, exista una lesión estructural en la neuroimagen, el niño tenga un déficit neurológico5,13 o haya actividad epileptógena clara en el EEG13.
Una vez que se decide iniciar tratamiento, lo ideal es la monoterapia5,13,21, ya que controla la CE en la mayoría de los niños, facilita el cumplimiento, minimiza los efectos adversos y evita las interacciones medicamentosas.
¿Qué fármaco antiepiléptico utilizar?
El FAE lo vamos a elegir en función del tipo de CE y de síndrome epiléptico, de la coomorbilidad, de los otros fármacos que tome (no olvidar que se reduce la efectividad de los anticonceptivos orales con los FAE inductores enzimáticos, como por ejemplo la carbamazepina), del estilo de vida (no es lo mismo un adolescente que un lactante) y de las preferencias del niño y su familia o cuidadores13.
En las tablas 3, 4 y 5 podemos ver resumidas las recomendaciones realizadas por las guías actuales después de analizar la evidencia científica disponible.
Tabla 4. Mostrar/ocultar
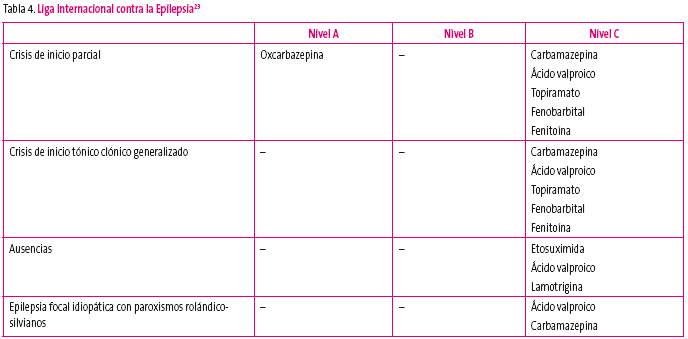
Tabla 5. Mostrar/ocultar
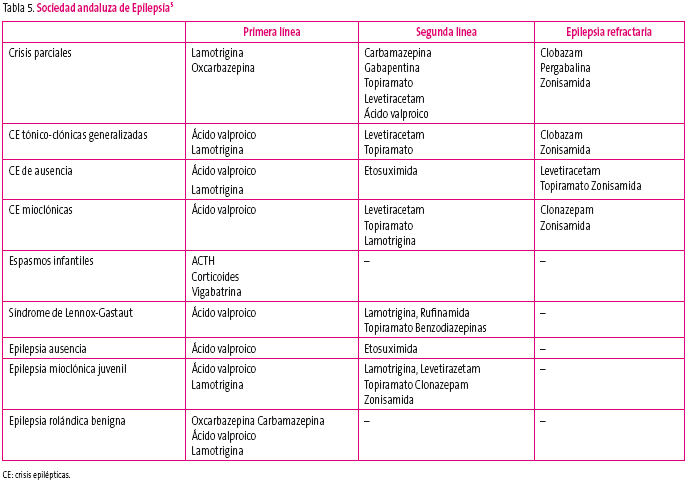
Debemos tener en cuenta que la etosuximida no es útil para las crisis tónico-clónicas, lo cual limita mucho su uso, aunque es un fármaco excelente para las ausencias, y que la lamotrigina puede empeorar las mioclonías en las epilepsias generalizadas primarias13. La vigabatrina es la primera opción si el síndrome de West se debe a una esclerosis tuberosa5,13.
En la práctica clínica habitual, como primera opción para las crisis y epilepsias generalizadas solemos utilizar el ácido valproico, por su amplio espectro clínico, la rapidez de su titulación y la facilidad de su uso al disponer de solución oral, comprimidos y presentación parenteral. Estas tres características las comparte con el levetiracetam y muy posiblemente son el motivo de que su uso esté aumentando, ya que sus competidores en este grupo de epilepsias (topiramato y lamotrigina) tienen una titulación lenta y carecen de presentación parenteral y de solución oral.
En el caso de las crisis o epilepsias focales, la oxcarbazepina es muy utilizada, no solo por su nivel de evidencia, sino por su fácil manejo (tiene solución oral y una titulación más rápida que otros, por ejemplo, la lamotrigina).
También debemos saber que algunos antiepilépticos deben evitarse en determinadas epilepsias: Lennox-Gastaut (carbamazepina y fenitoína), epilepsia ausencia infantil y juvenil (carbamazepina, oxcarbazepina, fenobarbital, tiagabina, vigabatrina y fenitoína), epilepsia mioclónica juvenil (carbamazepina, oxcarbazepina, gabapentina, tiagabina, vigabatrina y fenitoína)5,13.
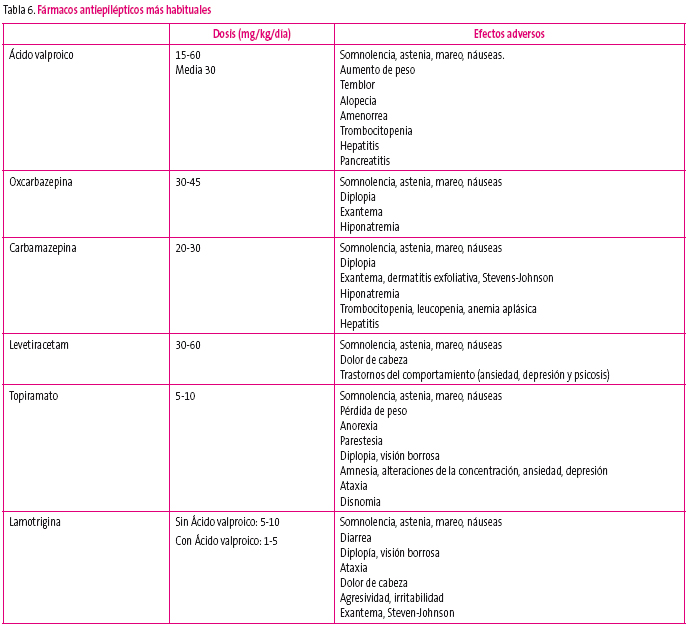
En la tabla 6 podemos ver las dosis recomendadas y los efectos adversos más habituales de los FAE más utilizados.
Tabla 6. Mostrar/ocultar
Recomendaciones generales
- Evitar situaciones en las que una pérdida de conocimiento pueda ser peligrosa: son cosas que todos deberíamos hacer. Ducha y no baño, acudir a la sierra y a la piscina acompañados, no ponerse en el borde del andén o la acera, etc.
- Ritmo de sueño adecuado: sobre todo para las epilepsias generalizadas idiopáticas.
- Abstención de bebidas alcohólicas y drogas: pensar que los adolescentes cada vez empiezan antes.
Crisis en un niño epiléptico conocido
Cuando el paciente diagnosticado de epilepsia sufre una nueva crisis, en primer lugar debemos asegurar que toma bien el tratamiento. Después preguntaremos por posibles situaciones en las que la absorción este disminuida (vómitos, diarrea, fármacos añadidos, etc.) y otros factores desencadenantes (por ejemplo, fiebre). Otra causa frecuente de descompensación es el aumento de peso; en este caso, reajustaremos la dosis del fármaco. Si disponemos de niveles terapéuticos del FAE utilizado nos ayudarán en este ajuste.
Los niveles los realizaremos siempre en valle y no desestimaremos un fármaco hasta que estemos en el límite alto o aparezcan afectos adversos.
Si no encontramos factores desencadenantes y la dosis del fármaco es la máxima posible, bien porque los niveles no nos permitan subir más, bien porque aparezcan efectos adversos o bien porque es la dosis máxima recomendada (en aquellos en los que no disponemos de niveles), deberemos plantearnos cambiar a otro FAE. La politerapia no es recomendable hasta haber realizado varios intentos de monoterapia5,13.
SEGUIMIENTO
En las revisiones habituales de todo paciente epiléptico debemos controlar el número y tipo de crisis e interrogar sobre los posibles efectos adversos.
Necesidad de analíticas
En el niño bien controlado no están indicadas de forma rutinaria las analíticas de control, incluyendo los niveles terapéuticos5.
Sí está indicado un hemograma y una bioquímica que incluya iones, perfil hepático y renal antes del inicio del tratamiento y al mes o a los seis meses, según los fármacos, fundamentalmente para descartar la leucopenia con la carbamazepina, la trombopenia y la hipertransaminasemia con el ácido valproico, y la hiponatremia con la carbamazepina y la oxcarbazepina. No debemos retirar el tratamiento salvo cuando los leucocitos bajen de 2000/mm3, los neutrófilos de 1000/mm3 o las plaquetas de 100 000/mm3. En el caso de las transaminasas, únicamente cuando triplican la normalidad5.
Necesidad de otras pruebas diagnósticas
La repetición del EEG no aporta información relevante cuando el diagnóstico no ofrece dudas. La repetición de la neuroimagen solo está indicada cuando existe un empeoramiento inexplicable de los síntomas neurológicos o cuando se plantea la cirugía de la epilepsia5.
Retirada del tratamiento
Después dos años sin crisis podemos plantear a los padres, y a nuestros pacientes si son lo suficientemente mayores, la retirada del tratamiento anticomicial13, aunque el riesgo de recurrencia después de la retirada se encuentra entre el 20 y el 30% (continuar con él tampoco asegura la ausencia de crisis)5. El riesgo de recurrencia es mayor en las epilepsias sintomáticas y si el EEG previo a la retirada es patológico5.
SÍNDROMES EPILÉPTICOS MÁS FRECUENTES EN LA INFANCIA
Epilepsia benigna de la infancia o epilepsia focal idiopática22
Epilepsia benigna de la infancia con paroxismo rolándico-silvianos
Muy frecuente, supone el 23% de las epilepsias de inicio en la edad escolar. Ocurre entre los 3 y los 13 años, con un pico a los 8-9. En el 65% de los casos las crisis ocurren al inicio del sueño o poco antes de despertar, aunque hay niños que sufren crisis diurnas y otros diurnas y nocturnas. El inicio de las CE es sensitivo unilateral en una hemilengua, labio, carrillo y faringe, en ocasiones con clonismos de la hemicara, anartria e hipersialorrea, rara vez generaliza. Suelen ser crisis muy breves (segundos o minutos). El EEG es característico: puntas de gran amplitud difásicas seguidas de onda lenta en la región rolándica. El excelente pronóstico (98-99,8% libres de crisis a los 18 años), el hecho de que suelan tener pocas crisis y ser focales y nocturnas hace que, en general, siempre de acuerdo con la familia, no se trate, salvo que haya muchas crisis o sean generalizadas.
Epilepsia benigna de la infancia con paroxismos occipitales
Se reconocen dos tipos, la de inicio precoz o síndrome de Panayiotopoulos (6-13% de las epilepsias focales) y la de inicio tardío o tipo Gastaut (0,15% de las epilepsias focales). En el primer tipo, las crisis tienen un componente autonómico importante, con palidez, cianosis perioral, hipersialorrea y vómitos, afectación de la conciencia, desviación tónica de la mirada y en ocasiones de la cabeza, y entre el 30 y el 50% progresa a una hemiconvulsión. Suelen ser crisis largas y no es raro que evolucionen a estatus; la edad de comienzo más habitual son los cinco años (2-12). En el tipo Gastaut, la edad más frecuente son los ocho años (3-16); las crisis suelen ser diurnas y durar menos de cinco minutos, el niño refiere pérdida de visión o alucinaciones visuales y se pueden generalizar. El EEG muestra puntas de gran voltaje seguidas de onda lenta en la región occipital en ambos tipos. En cuanto al tratamiento, también hay diferencias: la mitad de los niños con una forma precoz sufren una crisis única, por lo que en la mayoría de las ocasiones podremos no tratar; en cambio, en la forma de inicio tardío las crisis suelen ser muy frecuentes y, por tanto, sí se deben tratar.
Epilepsias generalizadas idiopáticas23
Suponen el 20% de las epilepsias de inicio en la infancia. Son niños intelectualmente normales que sufren crisis generalizadas y tienen descargas de punta-onda generalizada a más de 2,5 Hz en su EEG.
Epilepsia ausencia infantil
Inicio entre los 3 y los 12 años. Ausencias breves (5-15 segundos) con o sin parpadeo, mioclonías palpebrales o automatismos orofaciales. Lo más característico es que son muy numerosas en el día (hasta más de 100) y su recuperación es completa. La hiperventilación provoca ausencias. Hasta un 40% de los niños pueden tener crisis tónico-clónicas. El EEG es muy característico, con descargas de punta-onda generalizadas a 3Hz. El ácido valproico es el fármaco de elección ,ya que la etoxusimida no cubre las crisis tónico-clónicas. Después de un año con control absoluto (incluyendo EEG) puede plantearse la retirada del tratamiento, repitiéndose el EEG 6-8 semanas después de la retirada.
Epilepsia ausencia juvenil
Inicio algo más tardío y con menor número de ausencias al día. Las crisis tónico-clónicas aparecen en el 80% de los casos. La frecuencia de las descargas en el EEG suele ser de 4-5 Hz. El tratamiento de elección también es el ácido valproico, y la duración del mismo, más prolongada.
Epilepsia mioclónica juvenil
Inicio entre los 12 y los 18 años. Las crisis más características son las mioclonías en los miembros superiores al despertar (“tiran el desayuno”). El 90% sufre crisis tónico-clónicas, en general también al despertar, y precedidas de mioclonías. Algunos pacientes también tienen ausencias. Empeoran con la privación de sueño. La frecuencia de las descargas en el EEG también es mayor a 3 Hz y es frecuente la fotosensibilidad. El tratamiento de elección vuelve a ser el ácido valproico pero, dado el alto índice de recurrencias, se recomienda que se mantenga muchos años y en ocasiones de por vida.
Epilepsia con crisis tónico-clónicas al despertar
Inicio entre los 6 y los 25 años, las crisis características son las tónico-clónicas al despertar del sueño, aunque algunos pacientes las tienen también sin relación con el sueño. Pueden tener mioclonías o ausencias. La falta de sueño y el alcohol también son desencadenantes. El número de crisis suele ser pequeño a lo largo de la vida, pero el riesgo de recurrencia es alto.
Otras
Ausencias con mioclonías periorales, mioclonías palpebrales con ausencia.

LECTURAS RECOMENDADAS
-
National Clinical Guideline Centre. The Epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care [en línea]. London: National Clinical Guideline Centre. Available from: [www.nice.org.uk]. Updated 2011. Available from: http://www.nice.org.uk/nicemedia/live/12108/50164/50164.pdf.
Amplísima y excelente guía de práctica clínica realizada en el Reino Unido, que analiza de forma exhaustiva la evidencia científica disponible en 2011, modificando la previa del 2004- Existe una versión abreviada de la misma. -
Sociedad Andaluza de Epilepsia. Guía Andaluza de Epilepsia 2009. Diagnóstico y tratamiento de la epilepsia en niños y adultos. [en línea] [acceso 2011 Oct 11]. Disponible en: http://www.guiasalud.es/GPC/GPC_346_Andaluza_Epilepsia.pdf
Excelente y detallada guía elaborada por la Sociedad Andaluza de Epilepsia, que forma parte de las Guías de Práctica Clínica del Sistema Nacional de Salud español y que desmenuza la evidencia científica disponible de forma extensa.
BIBLIOGRAFÍA
- Hauser WA. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994;35(Suppl 2):S1-6.
- Cowan LD. The epidemiology of the epilepsies in children. Ment Retard Dev Disabil Res Rev. 2002;8(3):171-81.
- Durá Travé T, Yoldi Petri ME, Gallinas Victoriano F. Incidence of epilepsy in 0-15 year-olds. An Pediatr (Barc). 2007;67(1):37-43.
- Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. 2005;46(4):470-2.
- Sociedad Andaluza de Epilepsia. Guía Andaluza de Epilepsia 2009. Diagnóstico y tratamiento de la epilepsia en niños y adultos [en línea] [citado 2011 Oct 11].
- Proposal for revised clinical and electroencephalographic classification of epileptic seizures. From the Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1981;22(4):489-501.
- Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1989;30(4):389-99.
- Engel J Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42(6):796-803.
- Shorvon SD. The etiologic classification of epilepsy. Epilepsia. 2011;52(6):1052-7.
- Serrano-Castro PJ, Sánchez-Álvarez JC, Cañadillas-Hidalgo FM, Galán-Barranco JM, Moreno-Alegre V, Mercadé-Cerdá JM. Consensus clinical practice guidelines of the Sociedad Andaluza de Epilepsia for the diagnosis and treatment of patients with their first epileptic seizure in emergencies. Rev Neurol. 2009;48(1):39-50.
- Hirtz D, Ashwal S, Berg A, Bettis D, Camfield C, Camfield P et al. Practice parameter: evaluating a first nonfebrile seizure in children: report of the quality standards subcommittee of the American Academy of Neurology, The Child Neurology Society, and The American Epilepsy Society. Neurology. 2000;55(5):616-23.
- Guidelines for diagnosis and management of childhood epilepsy. Indian Pediatr. 2009;46(8):681-98.
- National Clinical Guideline Centre. The Epilepsies: the diagnosis and management of 28 the epilepsies in adults and children in primary and seconday care [en línea]. London: National Clinical Guideline 29 Centre. Available from: [www.nice.org.uk] 30 31 Updated 2011. Disponible en: http://www.nice.org.uk/nicemedia/live/12108/50164/50164.pdf
- Borusiak P, Zilbauer M, Jenke ACW. Prevalence of epileptiform discharges in healthy children--new data from a prospective study using digital EEG. Epilepsia. 2010;51(7):1185-8.
- Gaillard WD, Chiron C, Cross JH, Harvey AS, Kuzniecky R, Hertz-Pannier L et al. Guidelines for imaging infants and children with recent-onset epilepsy. Epilepsia. 2009;50(9):2147-53.
- Riviello JJ J.r, Ashwal S, Hirtz D, Glauser T, Ballaban-Gil K, Kelley K et al. Practice parameter: diagnostic assessment of the child with status epilepticus (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2006;67(9):1542-50.
- Zawadzki L, Stafstrom CE. Status epilepticus treatment and outcome in children: what might the future hold? Semin Pediatr Neurol. 2010;17(3):201-5.
- Sreenath TG, Gupta P, Sharma KK, Krishnamurthy S. Lorazepam versus diazepam-phenytoin combination in the treatment of convulsive status epilepticus in children: a randomized controlled trial. Eur J Paediatr Neurol. 2010;14(2):162-8.
- McMullan J, Sasson C, Pancioli A, Silbergleit R. Midazolam versus diazepam for the treatment of status epilepticus in children and young adults: a meta-analysis. Acad Emerg Med. 2010;17(6):575-82.
- Hirtz D, Berg A, Bettis D, Camfield C, Camfield P, Crumrine P et al. Practice parameter: treatment of the child with a first unprovoked seizure: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2003;60:166-75.
- Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C et al. ILAE treatment guidelines: evidence-based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2006;47:1094-120.
- Chahine LM, Mikati MA. Benign pediatric localization-related epilepsies. Epileptic Disord. 2006;8(4):243-58.
- Camfield C, Camfield P. Management guidelines for children with idiopathic generalized epilepsy. Epilepsia. 2005;46(Suppl 9):112-6.