


Aumento aislado de transaminasas: aproximación diagnóstica
2 CS Cuzco. Fuenlabrada. Madrid (España).
RESUMEN: PUNTOS CLAVE PARA EL PEDIATRA DE ATENCIÓN PRIMARIA
SUMMARY: KEY POINTS FOR PRIMARY CARE PEDIATRICIANS
PUNTOS CLAVE
- La hipertransaminasemia aislada como hallazgo casual en un paciente asintomático es relativamente frecuente.
- Puede ser la primera manifestación de una hepatopatía potencialmente grave o la manifestación de enfermedades extrahepáticas.
- La ausencia de síntomas no asegura la benignidad del proceso.
- Es necesario realizar una reevaluación de la historia y de la exploración del paciente buscando datos que hubieran pasado desapercibidos.
- La hipertransaminasemia debe ser confirmada y seguida hasta su normalización.
- En ciertas patologías, el diagnóstico etiológico de la misma puede prevenir el desarrollo de clínica y secuelas irreversibles y, en su caso, permitir la realización de consejo genético.
INTRODUCCIÓN
La hipertransaminasemia o elevación de aminotransferasas como alteración analítica aislada detectada de forma casual es un hallazgo relativamente frecuente en el paciente pediátrico. Es un dato inespecífico que puede ser la primera manifestación de una hepatopatía potencialmente grave en un paciente asintomático, o deberse a procesos extrahepáticos.
El hígado interviene en numerosos procesos metabólicos, de producción, almacenaje y eliminación que afectan a hidratos de carbono, lípidos, proteínas, vitaminas, hormonas y sustancias endógenas como la bilirrubina o exógenas como fármacos, y en él tiene lugar la síntesis de factores imprescindibles como los factores de coagulación.
En la práctica clínica utilizamos numerosas determinaciones analíticas, que evalúan distintos aspectos de la funcionalidad hepática y que informan sobre:
- Citolisis o lesión hepatocelular: transaminasas, aspartato-aminotransferasa o transaminasa glutámico oxalacética (AST o GOT) y alanino-aminotransferasa o transaminasa glutámico pirúvica (ALT o GPT).
- Colestasis o éstasis biliar y metabolismo de la bilirrubina: Fosfatasa alcalina, gamma glutamil traspeptidasa (GGT), bilirrubina total y fraccionada.
- Capacidad de síntesis: albúmina y coagulación1.
En este artículo haremos referencia fundamentalmente a la elevación sérica de transaminasas, AST y ALT. Son enzimas que catalizan la transferencia reversible del grupo α-amino de los aminoácidos alanina y ácido aspártico al grupo α-ceto del ácido cetoglutárico. Son intracelulares, ALT se localiza en el citosol y AST en el citosol (20%) y en la mitocondria (80%); se liberan hacia la sangre en grandes cantidades cuando hay daño en la membrana de las células que la contienen.
La AST, además de en el hepatocito, se encuentra en otros tejidos, corazón, músculo esquelético, riñones, cerebro, páncreas, pulmón, eritrocitos y leucocitos, mientras que la ALT se localiza fundamentalmente en el hepatocito y en mucha menor cantidad que en otros tejidos, siendo por tanto más específica de daño hepático.
Concepto de hipertransaminasemia
Se considera hipertransaminasemia el aumento del valor sérico de las transaminasas por encima de las cifras normales según el laboratorio de referencia. El límite superior suele estar alrededor de 40-45, aunque es superior en el primer año de la vida2.
Los valores séricos de transaminasas pueden variar en condiciones normales en función del laboratorio, de las condiciones de extracción de la muestra, de la edad, el sexo y la raza del paciente, de la realización de ejercicio previo, de la hemólisis, del daño muscular o de un traumatismo previo1; por ello, ante una alteración de los mismos siempre deberá repetirse la determinación y confirmar el dato.
La causa más importante del aumento de la ALT sérica es la enfermedad hepática, bien debida a una lisis de los hepatocitos o a una alteración transitoria de la permeabilidad de la membrana. La enfermedad hepática es también una causa frecuente del aumento de AST sérica.
Debe analizarse con precaución la relación entre niveles de la alteración y gravedad y pronóstico de la causa que la motiva, pues puede haber niveles normales o muy levemente elevados en hepatitis crónica y cirrosis. Igualmente, el descenso de cifras previamente elevadas no siempre implicará mejoría, ya que a veces un descenso rápido y brusco, acompañado de una elevación de bilirrubina y un alargamiento del tiempo de protrombina, puede objetivarse en el contexto de una necrosis hepática submasiva grave. No obstante, distintos autores establecen una aproximación diagnóstica según si el grado de la elevación es superior o inferior a diez veces los valores normales1,3.
Frecuencia
En adolescentes estadounidenses de edades entre 12 y 19 años se objetiva aumento aislado de transaminasas en un 7,4% de la población4, siendo más frecuente en niños que en niñas.
ETIOLOGÍA. DIAGNÓSTICO DIFERENCIAL
La etiología varía en función de la muestra estudiada, su edad, las condiciones de exclusión, los medios diagnósticos utilizados y el tiempo de seguimiento5. En una serie de 259 niños de 1 a 18 años de edad en los que se objetiva un aumento de transaminasas de forma casual con una duración inferior a los seis meses y en los que se había excluido hepatitis por virus A, B y C, la causa en más del 50% de los casos era infecciosa, secundaria a la infección por virus de Epstein-Barr, citomegalovirus y toxoplasma. En el 5%, de los casos el origen estaba en la ingesta previa de fármacos y en un 43% no se identificó ninguna causa. En los 166 pacientes en los que la alteración persistió más de seis meses, se identifican como causas responsables de la misma y por orden de frecuencia: obesidad, enfermedad de Wilson, distrofia muscular, hepatitis autoinmune, alteraciones biliares (colelitiasis y quistes de colédoco), síndrome de Alagille, intolerancia hereditaria a la fructosa, glucogenosis tipo IX, enfermedad celíaca, déficit de ornitina trascarbamilasa y síndrome de Shwachman. Llama la atención a los autores el alto número de enfermedades genéticas, hasta ese momento asintomáticas, identificadas en el estudio. En un 13% de los pacientes no se pudo identificar la causa de la hipertransaminasemia antes de la realización de biopsia6.
Por tanto, hay que considerar, incluso en el paciente asintomático, que puede ser el primer dato de una enfermedad grave que es preciso identificar para instaurar el tratamiento adecuado que permita evitar el desarrollo de complicaciones y secuelas o, en su caso, realizar el consejo genético pertinente.
Así, ante una elevación aislada de las cifras de transaminasas hay que hacer un diagnóstico diferencial amplio que incluya:
- Causas hepáticas.
- Causas extrahepáticas.
- Normalidad.
Entre las entidades más frecuentemente responsables de la elevación de transaminasas hay que considerar:
Infecciones
Puede observarse elevación de transaminasas en procesos infecciosos transitorios, tanto sistémicos como locales, fundamentalmente en lactantes y niños pequeños y con elevaciones que no superan el doble del límite superior de los valores normales. Puede tratarse de infecciones respiratorias o gastrointestinales generalmente debidas a virus (adenovirus, virus respiratorio sincitial, parvovirus y herpesvirus). En lactantes hay que considerar la posibilidad de una infección urinaria debidas a Escherichia coli, por lo que en el estudio inicial se debe incluir un urocultivo. En niños más mayores, los agentes responsables pueden ser virus de Epstein-Barr, citomegalovirus, virus de la varicela-zóster, bacterias como Salmonella o parásitos como toxoplasma7.
Las hepatitis virales (A, B, C, D y E) actualmente son poco frecuentes en nuestro medio tras la mejora en las condiciones higiénicas; la inmunización universal contra hepatitis B y en situaciones de riesgo contra hepatitis A, y el control de la transmisión vertical y de las fuentes de transmisión parenteral. Sin embargo, deben tenerse en cuenta en niños no inmunizados, nacidos de embarazo no controlado, de los que no se conocen datos o que provienen de zonas endémicas por origen o viajes recientes. Cualquiera de las hepatitis mencionadas puede cursar de forma asintomática. En el caso de la hepatitis A, nunca puede atribuirse a esta etiología una elevación de transaminasas superior a los seis meses.
Ingesta de fármacos y sustancias hepatotóxicas
Hay que valorar la ingesta de fármacos potencialmente hepatotóxicos, algunos tan frecuentemente utilizados como la amoxicilina/ácido clavulánico o los macrólidos, pero también de otros que pueden pasar inadvertidos tales como productos de herbolario u homeopáticos, así como el uso de sustancias para aumentar el rendimiento deportivo8,9. En niños más mayores y adolescentes debe considerarse el consumo de tóxicos como alcohol o drogas (Tabla 1).
Tabla 1. Mostrar/ocultar
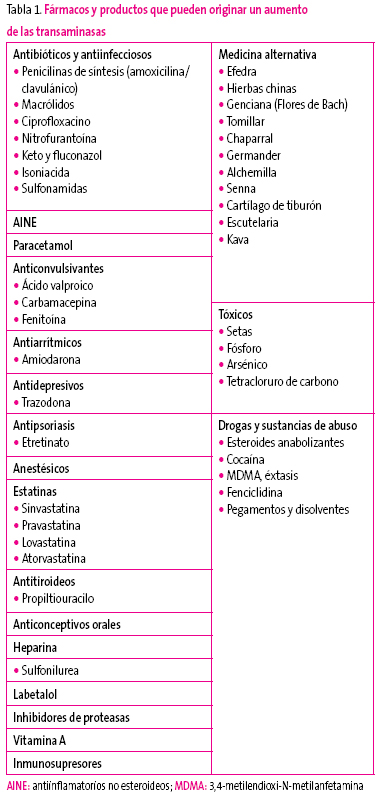
Obesidad
En adolescentes obesos y con sobrepeso se observa elevación de transaminasas con mayor frecuencia que en la población general10 en relación con una prevalencia de hígado graso en obesos que alcanza el 38%, mientras que solo es del 5% en chicos con normopeso11.
Hepatitis autoinmune
La hepatitis autoinmune es una enfermedad hepática inflamatoria progresiva que cursa con aumento de transaminasas y de inmunoglobulina IgG, autoanticuerpos positivos y afectación histológica hepática en ausencia de etiología conocida. Se divide en dos tipos: tipo I, con anticuerpos antimúsculo liso y/o antinucleares, y tipo II, con anticuerpos antimicrosomales de hígado y riñón. Ambos son más frecuentes en niñas. Un 10-15% de los pacientes puede no presentar estos anticuerpos al diagnóstico. En el momento del diagnóstico, los pacientes pueden ser asintomáticos y presentar solo alteraciones analíticas; sin embargo, es una enfermedad grave y de curso progresivo que también puede presentarse como hepatitis fulminante o cirrosis. Puede asociarse con otras enfermedades autoinmunes como tiroiditis, diabetes mellitus, anemia hemolítica, enfermedad celíaca o colitis ulcerosa. Ante su sospecha, sobre todo en niñas, aunque no exclusivamente, con aumento intenso de transaminasas y con serología negativas, debe derivarse a la paciente a un Servicio de Gastroenterología Infantil con objeto de no demorar su diagnóstico y tratamiento que se realiza con corticoides a los que pueden añadirse inmunosupresores (prednisona/azatioprina). Puede requerir incluso trasplante hepático12,13.
Enfermedad de Wilson
Es un trastorno autosómico recesivo causado por una mutación en el gen ATP7B. Se produce una alteración en el metabolismo del cobre que se acumula en distintos tejidos y órganos. Cursa con alteraciones neurológicas, psiquiátricas y del comportamiento, hepatopatía, anemia hemolítica, aparición de anillo de Kayser-Fleischer por depósito de cobre en la córnea y, más raramente, afectación renal semejante al síndrome de Fanconi, artritis e hipoparatiroidismo. La hepatopatía incluye hepatomegalia asintomática, hepatitis subaguda y crónica, cirrosis e insuficiencia hepática fulminante. El tipo de mutación se asocia con la edad de inicio, que puede ser tan precoz como los dos o tres años. En pacientes más jóvenes, es más probable que predomine la afectación hepática. Para el diagnóstico se debe medir la ceruloplasmina sérica, disminuida en el 85% de los casos, y la eliminación de cobre en orina de 24 horas. El diagnóstico de certeza lo establece la biopsia hepática, que también determina la gravedad y extensión de la afectación hepática. El tratamiento, además de restringir el cobre de la dieta, es la utilización de quelantes de cobre, penicilamina, trientina y acetato de zinc, para eliminar su exceso y evitar su depósito en los tejidos. Por este motivo, es importante diagnosticar precozmente la enfermedad14,15.
Déficit de α1 antitripsina
Es una enfermedad genética con herencia autosómica dominante. Se caracteriza por la imposibilidad del hígado para secretar α1 antitripsina (A1AT) a la sangre y su acúmulo en dicho órgano. La A1AT es un inhibidor de la proteasa, que protege el tejido alveolar pulmonar de la elastasa de los neutrófilos y los irritantes inhalados. Su déficit puede cursar con colestasis neonatal y lesión hepática variable hasta cirrosis, así como enfisema pulmonar más tardíamente. El alelo más frecuente del sistema inhibidor de la proteasa (Pi) es M y el fenotipo normal es PiMM. La clínica es variable dependiendo de los alelos existentes, y no siempre cursa con hepatopatía. Se debe sospechar ante niveles bajos de A1AT sérica y ausencia de un pico en las α-globulinas en la electroforesis. Se debe completar el estudio con el análisis de fenotipos (M, S y Z)14,15.
Hemocromatosis
Es una enfermedad autosómica recesiva, con mutaciones puntuales en el gen HFE, que se manifiesta en la adolescencia y la edad adulta. Existe un cuadro neonatal con siderosis hepática y extrahepática que puede tener un curso fulminante. Se ve afectado el metabolismo del hierro, provocando un acúmulo excesivo de este en los órganos. Debe sospecharse ante una saturación de transferrina >45% y ferritina aumentada. Según los datos analíticos y el estudio genético puede estar indicada la realización de biopsia hepática. En hijos de afectados debe realizarse estudio genético e iniciar seguimiento si este es compatible con el diagnóstico14,15.
Enfermedad celíaca
Se trata de una enfermedad sistémica inmunomediada provocada por ingesta de gluten en individuos genéticamente susceptibles. Se ha descrito una amplia variedad de afectaciones hepáticas incluida la elevación de transaminasas sin otros síntomas y previa al diagnóstico de la enfermedad. Esta elevación desaparece con la dieta sin gluten16.
Miopatías
Dada la presencia de AST, y en menor cantidad de ALT en células musculares, en situaciones en que se produce daño muscular tal como ejercicio intenso o enfermedades musculares puede observarse elevación de las mismas, particularmente AST. Esto incluye errores del metabolismo, miopatías congénitas y adquiridas. En estos casos se produce elevación de aldolasa y creatinfosfoquinasa, así como la aparición de mioglobinuria. Como se describe en la literatura, la hipertransaminasemia mantenida puede ser la forma de debut de miopatías en niños considerados asintomáticos.
Hiper-/hipotiroidismo
Se ha descrito aumento de transaminasas tanto en hiper como en hipotiroidismo. No está claro el mecanismo de producción, podría deberse a afectación muscular en el hipotiroidismo y a un efecto directo tirotóxico. También es posible por el empleo de fármacos antitiroideos.
También se ha descrito en adultos en insuficiencia suprarrenal y en anorexia nerviosa.
Macrotransaminasemia
La macro aspartato aminotransferasa es una macroenzima, un complejo de alto peso molecular formado por AST con otros componentes del plasma. Aparece más frecuentemente en adultos y asociada a enfermedades neoplásicas o autoinmunes, pero puede verse también en niños, en los que es más frecuentemente un proceso benigno y puede explicar hasta un tercio de casos con elevación exclusiva de AST. Como cribado diagnóstico, puede utilizarse el porcentaje de AST precipitada con polietilenglicol (%PPA) y el diagnóstico se confirma con electroforesis. También se ha asociado con enfermedad inflamatoria intestinal17.
Otras causas
Además de estas etiologías, hay que considerar otras causas menos frecuentes como3,6,18:
- Trauma obstétrico, cromosomopatías.
- Fibrosis quística del páncreas.
- Intolerancia a las proteínas de leche de vaca.
- Metabolopatías (glucogenosis, galactosemia, fructosemia) porfirias.
- Enfermedades de depósito (Gaucher, Nieman Pick).
- Enfermedades hepatobiliares: colestasis neonatal intrahepática, atresia biliar, quiste de colédoco, síndrome de Alagille.
- Nutrición parenteral prolongada (con datos de colestasis).
- Neoplasias (hepatoblastoma y neuroblastoma).
- Síndrome de Budd Chiari.
- Colagenosis.
ORIENTACIÓN DIAGNÓSTICA
En un paciente con aumento aislado de aminotransferasas sin otra sintomatología se debe realizar una detallada anamnesis y una exploración física cuidadosa, que permita orientar el diagnóstico, solicitándose posteriormente pruebas de laboratorio y exploraciones complementarias escalonadas.
Anamnesis
Debe ser acorde con la edad del paciente, debe incluir antecedentes personales desde el embarazo, controlado o no, parto y periodo neonatal, posibilidad de traumatismo previo, transfusiones y cirugías, adquisición de hitos motores, vacunaciones recibidas, enfermedades previas, realización de ejercicio, ingesta de fármacos, incluidos complejos vitamínicos y preparados de herbolario, hábitos tóxicos o viaje internacional reciente, lugar de nacimiento y residencia, y antecedentes familiares de enfermedades metabólicas y autoinmunes, tales como hepatitis autoinmune, tiroiditis y diabetes mellitus e infecciones en la familia y entorno.
Debe preguntarse por existencia de coluria, acolia, prurito y sangrado.
Exploración física
En un paciente en el que la elevación de transaminasas se detecta de forma casual es raro el hallazgo de signos en la exploración; aun así, se debe realizar una búsqueda activa de los mismos. Se debe considerar la existencia de ictericia, distensión y dolor abdominal, existencia de hepato- y esplenomegalia y signos de sangrado. Valorar el estado de nutrición y el índice de masa corporal (IMC) para evaluar la repercusión que podría tener una enfermedad crónica o identificar un estado de obesidad responsable de la alteración. Buscar dismorfias faciales, fenotipo peculiar como en el síndrome de Alagille, alteraciones oculares (enfermedad de Wilson) o alteraciones del desarrollo psicomotor que orientarían a metabolopatías. Si existe hepatopatía crónica, se podrían evidenciar arañas vasculares, eritema palmar, xantomas o acropaquias. También se deben explorar la marcha y el tono muscular.
En la Tabla 2 figura un esquema con los datos que deben recogerse en estos pacientes.
Tabla 2. Mostrar/ocultar
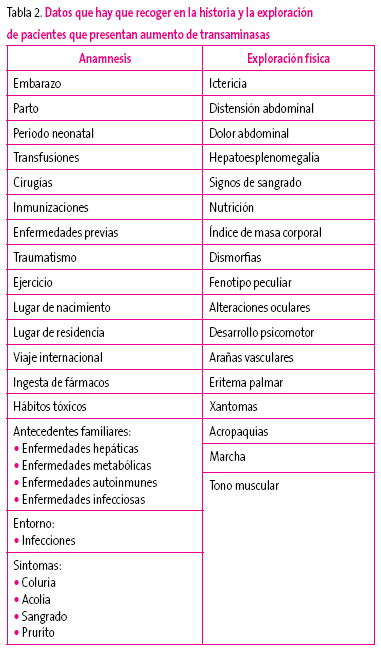
Si de los datos de la historia y/o la exploración se identifica una causa que pudiera ser la responsable de la hipertransaminasemia y sobre la que pudiera intervenirse, debe procederse a su eliminación.
Pruebas complementarias
Ante el hallazgo de una hipertransaminasemia asintomática detectada de forma casual hay que repetir la analítica en un plazo de 15-30 días para:
- Confirmar la alteración.
- Evaluar la función hepática con un perfil hepático completo Pruebas complementariascon marcadores de colestasis (GGT, FA, bilirrubina total y fraccionada), albúmina y coagulación.
- Hacer una aproximación etiológica. Si de los datos de la historia y/o la exploración se sospecha una patología determinada, deben realizarse las pruebas complementarias indicadas para su diagnóstico. De no ser así, se solicitará serología Pruebas complementariasde virus hepatotropos (virus de las hepatitis A, B y C, virus de Epstein Barr, citomegalovirus) y según datos de la historia en lactantes TORCH (toxoplasma, citomegalovirus, rubeola, lúes y virus de la inmunodeficiencia humana).
Además, en lactantes se debe realizar urocultivo para descartar infección urinaria.
En niños obesos se debe realizar una ecografía abdominal.
ALGORITMO DIAGNÓSTICO ESCALONADO
- Si tras esta valoración inicial se observara normalización de las cifras se debería constatar que esta normalización persiste en el tiempo al menos con una nueva determinación.
- Si se identificara una causa responsable de la alteración se procederá al tratamiento o seguimiento de la misma.
-
Si no se identificara causa responsable y la alteración persistiera debe realizarse en uno o dos meses un nuevo estudio ampliado que debe incluir la determinación de enzimas musculares (creatinfosfoquinasa y aldolasa), serología de enfermedad celíaca (anticuerpos antitrasglutaminasa e IgA), hormona tiroestimulante (TSH), índice de saturación de trasferrina (IST) y ferritina, alfa 1 antitripsina, ceruloplasmina e ionotest, así como ecografía abdominal, si no se realizó previamente, para valorar la morfología hepática y las vías biliares.
Puede determinarse la existencia de autoanticuerpos (anticuerpos antinucleares, anticuerpos antimúsculo liso y anticuerpos microsomales contra hígado y riñón) pero, dada la posibilidad de que, en ocasiones, la hepatitis autoinmune pueda presentarse con negatividad de estos autoanticuerpos, que a su vez estos puedan estar presentes en otras patologías, así como la necesidad de contar con un laboratorio especializado, es aconsejable que estas pruebas sean solicitadas e interpretadas por un gastroenterólogo pediátrico.
Ante sospecha de macrotransaminasemia se debe solicitar determinación de AST precipitada con polietinelglicol, si bien el diagnóstico definitivo se realiza con electroforesis.
Si se identificara una causa responsable de la alteración se procederá al tratamiento o seguimiento de la misma. - Si no se identificara causa responsable y la alteración persistiera debe realizarse seguimiento y continuación del proceso diagnóstico. Si persistiera durante seis meses sin causa filiada debe ser remitida a una Unidad de Gastroenterología y Hepatología Pediátrica.
- Si se detectara una elevación de transaminasas (x10) en dos determinaciones sin causa etiológica filiada, y sobre todo en pacientes de sexo femenino, la derivación debe realizarse sin demora ante la posibilidad de que se trate de una hepatitis autoinmune que pudiera beneficiarse de tratamiento precoz.
Recomendamos como guía diagnóstica el algoritmo diagnóstico que propone el grupo GastroSur3, orientado también según edad e intensidad de la elevación. En la Tabla 3 figuran las patologías que deben ser consideradas en el diagnóstico diferencial, junto con las pruebas complementarias indicadas en cada una de ellas.
Tabla 3. Mostrar/ocultar
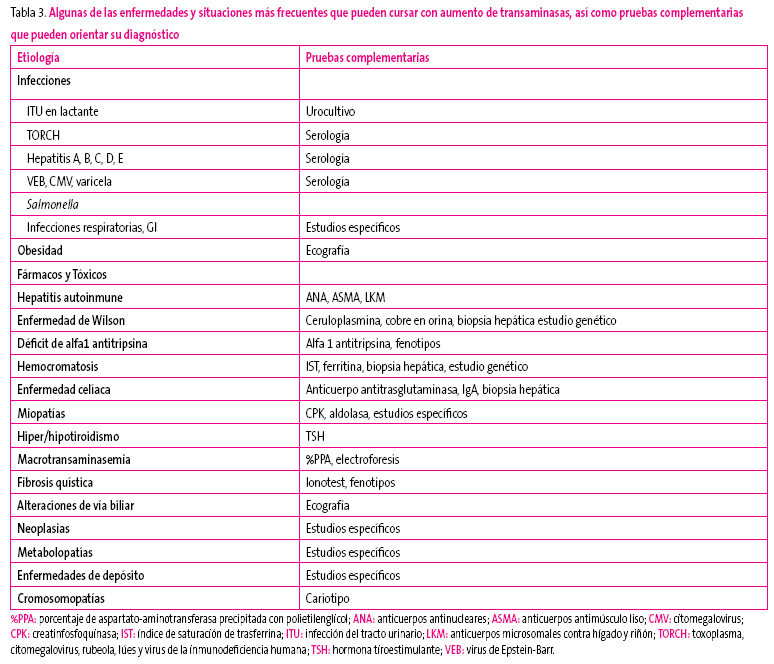
INDICACIONES DE DERIVACIÓN
Urgente
- Datos de hepatopatía grave: encefalopatía (no esperable en este contexto), disminución de protrombina, hipoglucemia o hipoalbuminemia importante.
- Datos de colestasis en lactantes pequeños.
Indicación de derivación para completar estudio
- Sospecha de hepatitis autoinmune.
- Datos asociados de enfermedad metabólica y/o neurológica.
- Colestasis en niños mayores.
- Cuando se realice el diagnóstico de enfermedades que precisen seguimiento especializado.
- En cualquier caso, si la elevación se prolonga más de seis meses.
LECTURAS RECOMENDADAS
- Albañil MR, Carabaño I, Galiano MJ, Guerra ME, Manzanares J, Medina E, et al. Hipertransaminasemia. Guías Conjuntas de Patología Digestiva Pediátrica Atención Primaria-Especializada [en línea]. Disponible en http://www.ampap.es/documentacion/protocolos/Hipertransaminasemia_2008.pdf
- Ofrece una aproximación clara y completa al tema, proponiendo una estrategia práctica de su abordaje.
- Giannini E, Testa R, Savarino V. Liver enzyme alteration: a guide for clinicians. CMAJ. 2005;172:367-79.
- Sin ser específicamente dirigido a población pediátrica, aporta un análisis muy útil para la comprensión del tema.
-
Iorio R, Sepe A, Gainnattasio A, Cirillo A, Vegnente a. Hypertransaminasemia in chilhood as a marker of genetic liver disease. J Gastroenterol. 2005;40:820-6.
Recoge una serie de casos con las etiologías responsables. Hay que tener en cuenta el contexto en el que se realiza.
BIBLIOGRAFÍA
- Giannini E, Testa R, Savarino V. Liver enzyme alteration: a guide for clinicians. CMAJ. 2005;172:367-79.
- Landaas S, Skrede S, Steen JA. The levels of serum enzymes, plasma proteins and lipids in normal infants and small children. J Clin Chem Clin Biochem. 1981;19:1075-80.
- Albañil MR, Carabaño I, Galiano MJ, Guerra ME, Manzanares J, Medina E, et al. Guías Conjuntas de Patología Digestiva Pediátrica Atención Primaria-Especializada [en línea] [consultado el 28/8/2012]. Disponible en: http://www.ampap.es/documentacion/protocolos/Hipertransaminasemia_2008.pdf
- Fraser A, Longnecker M, Lawlor D. Prevalence of elevated alanine aminotransferasa among US adolescents and associated factors: NHANES 1999-2004. Gastroenterology. 2007;133:1814-20.
- Bugeac N, Pacht A, Mandel H, Iancu T, Tamir A, Srugo I, et al. The significance of isolated elevation of serum aminotransferases in infants and young children. Arch Dis Child. 2007;92:1109-12.
- Iorio R, Sepe A, Giannattasio A, Cirillo A, Vegnente A. Hypertransaminasemia in chilhood as a marker of genetic liver disease. J Gastroenterol. 2005;40:820-6.
- Frauca E. Ictericia e hipertransaminasemia. Actitud diagnóstico-terapéutica. Rev Pediatr Integ. 2003;VII:187-203.
- Giboney P. Mildly elevated liver transaminase levels in the asymptomatic patient. Am Fam Physician. 2005;71:1105-10.
- Díaz A, De la Fuente S, Castiñeira C, Costa C. Hipertransaminasemia. Guías Clínicas Fisterra [en línea] [consultado el 28/8/2012]. Disponible en: http://www.fisterra.com/guias-clinicas/hipertransaminasemia/
- Strauss R, Barlow S, Dietz W. Prevalence of abnormal liver enzymes in obese and very obese adolescents. J Pediatr. 2000;136:727-33.
- Schwimmer J, Deutsch R, Kahen T, Lavine J, Stanley C, Behling C. Prevalence of Fatty Liver in Children and Adolescents. Pediatrics. 2006;118:1388-93.
- Makol A, Watt K, Chowdhary R. Autoimmune Hepatitis: A review of current, diagnosis and treatment. Hepat Res Treat. 2011;2011:390916.
- Mieli-Vergani G, Heller S, Jara P, Vergani D, Chang M, Fujisawa T, et al. Autoimmune hepatitis. JPGN. 2009;49:158-64.
- Kaplan M, Chopra S, Travis A. Approach to the patient with abnormal liver function tests. UpTodate [en línea] [consultado el 23/8/2012]. Disponible en: www.uptodate.com
- Carey R, Balistreri W. Enfermedades metabólicas del hígado. En: Kliegman RM, Stanton BF, St. Geme III JW, Schor NF (eds.). Nelson Tratado de Pediatría, Vol. II, 18.ª ed. Barcelona: Elsevier España; 2009. p. 1675-80.
- Abdo A, Meddings J, Swain M. Liver abnormalities in celiac disease. Clin Gastroenterol Hepatol. 2004;2:107-12.
- Caropreso M, Fortunato G, Lenta S, Palmieri D, Esposito M, Vitale D, et al. Prevalence and long-term course of macro-aspartate aminotransferase in children. J Pediatr. 2009;154:144-748.
- García M, Zurita A. Transaminasas: Valoración y significación clínica. Protocolos de la AEP. Protocolos de Gastroenterología, Hepatología y Nutrición. Asociación Española de Pediatría, Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica 2010, 2.ª ed. Madrid: Ergón; 2012. p. 267-75 [en línea]. Disponible en: http://www.aeped.es/sites/default/files/documentos/transaminasas.pdf