


Cribado neonatal de enfermedades endocrinometabólicas
2 J. García Aguado; M. Merino Moína; C. R. Pallás Alonso; J. Pericas Bosch; F. J. Sánchez Ruiz-Cabello; F. J. Soriano Faura; J. Colomer Revuelta; O. Cortés Rico; M. J. Esparza Olcina; J. Galbe Sánchez-Ventura; A. Martínez Rubio. (España).
RESUMEN: PUNTOS CLAVE PARA EL PEDIATRA DE ATENCIÓN PRIMARIA
SUMMARY: KEY POINTS FOR PRIMARY CARE PEDIATRICIANS
RESUMEN
Desde finales de la década de los 70, muchos países pusieron en marcha programas de cribado metabólico neonatal encaminados al diagnóstico precoz de aquellas metabolopatías congénitas que, detectadas en los primeros días de vida, fuesen susceptibles de tratamiento. De esta manera se mejoró notablemente el pronóstico de algunas de estas enfermedades. Inicialmente, los programas incluyeron el cribado de la fenilcetonuria y del hipotiroidismo congénito, siendo ampliados posteriormente con el de la hiperplasia suprarrenal congénita y de la fibrosis quística de páncreas. Recientemente, la introducción de la espectrometría de masas en tándem ha permitido el cribado de muchas metabolopatías; sin embargo, muchas de ellas no reúnen los criterios mínimos para ser incluidas en un programa de cribado universal.
PUNTOS CLAVE
- Es necesario que el pediatra de Atención Primaria compruebe que se ha realizado el cribado neonatal y que se ha registrado correctamente.
- La comunicación de una anomalía o de un posible diagnóstico de trastorno congénito del metabolismo debe producirse en los primeros 15 días de vida.
- Durante la punción del talón deben adoptarse medidas no farmacológicas para evitar el dolor, como el amamantamiento, el contacto piel con piel o las soluciones de sacarosa.
- Los padres deben ser informados, mejor si puede ser por escrito, de las pruebas de cribado metabólico neonatal.
- Ha sido un acuerdo importante entre el Ministerio de Sanidad y el Consejo Interterritorial la unificación de los programas de cribado metabólico neonatal mediante un único programa para todo el territorio español.
INTRODUCCIÓN
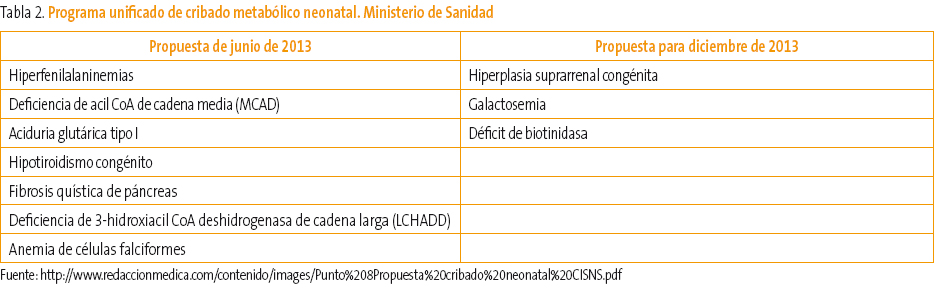
La detección sistemática de metabolopatías neonatales se inicia en España, al igual que en otros países de nuestro entorno, a finales de la década de los 70 del siglo XX. Desde entonces, la mayoría de los países desarrollados han introducido programas de detección de metabolopatías centrados fundamentalmente en el cribado del hipotiroidismo congénito (HC) y de la fenilcetonuria (FC). Según el informe realizado por J. Soriano Faura1 sobre la variabilidad de actividades preventivas realizadas en España en 2011, en cinco comunidades autónomas (CCAA) disponen de espectrometría de masas en tándem (EMT) y criban respectivamente 17, 19, 30 y 40 trastornos. En cinco CCAA se realizan tres cribados, en una se realizan dos, en dos CCAA cuatro trastornos y dos trastornos en otras seis. En la Tabla 1 podemos ver el número de niños cribados en España de las distintas metabolopatías1-3 hasta junio de 2013. Como vemos, la variabilidad del cribado neonatal es muy amplia a lo largo de toda la geografía española, a pesar de que los expertos indican que para una gestión eficiente de los recursos cada laboratorio de cribado debería procesar unas 50 000 muestras anuales. Sería deseable por tanto un programa único de cribado para todo el territorio español realizado con la mejor tecnología disponible (EMT) y coordinado entre todas las CCAA. Desde junio de 2013, el Ministerio de Sanidad y el Consejo Interterritorial han acordado un programa de cribado neonatal para todo el territorio español3. Son siete los trastornos que se consideran prioritarios y que pueden verse en la Tabla 2.
Tabla 1. Mostrar/ocultar
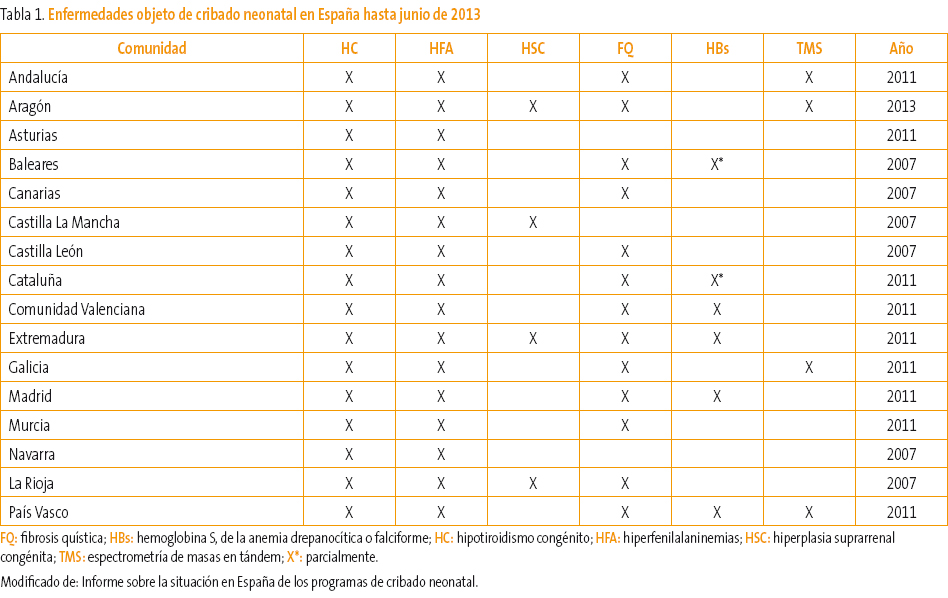
Tabla 2. Mostrar/ocultar
Los criterios que deben cumplirse para incluir en el cribado de errores innatos del metabolismo son los siguientes2-6:
- Debe tratarse de una anomalía relativamente frecuente, al menos 1:15 000 recién nacidos.
- Debe producir una grave anomalía metabólica.
- Debe ser difícil de diagnosticar clínicamente durante el periodo neonatal.
- El diagnóstico clínico debe producirse tras una fase preclínica asintomática.
- Debe existir un marcador bioquímico con una buena sensibilidad y especificidad, que permita discriminar a los recién nacidos sanos de los enfermos durante la fase preclínica.
- Debe ser posible realizar un tratamiento de la enfermedad de forma precoz, que mejore sensiblemente el pronóstico de la misma.
- El coste del programa de prevención debe ajustarse a los criterios de evaluación económica, como todo programa de salud pública.
Cualquier anomalía detectada debe ser conocida y comunicada en los primeros 15 días de vida. Se describen a continuación de forma resumida los trastornos considerados por los expertos como prioritarios para ser objeto de cribado5,6.
TRASTORNOS OBJETO DE CRIBADO NEONATAL
Hipotiroidismo congénito
El HC es causa de importantes alteraciones en el desarrollo cerebral del feto, produciendo anomalías estructurales permanentes. Se estima que solamente un 5% de los recién nacidos con HC presentará manifestaciones clínicas. Existen varios genes sospechosos del trastorno, como los TTF1, TTF2, PAX8 y TSHR locus 2q12-q I 4. La prevalencia en España de de 1:2344. Es más frecuente en el sexo femenino (2:1). La herencia es autosómica recesiva. La clínica consiste en retardo mental, retraso de crecimiento, macroglosia, piel áspera y hernia umbilical2,4-6.
Prueba de cribado
Existen dos estrategias:
- Determinación primaria de hormona tiroestimulante (TSH) si esta supera los 10 µ/ml y es menor de 25 µ/ml se realiza una segunda determinación. Si la TSH supera los 25 µ/ml se realiza la confirmación diagnóstica.
- Basada en la determinación de T4 con un punto de corte de 6 µ/ml y de TSH punto de corte 25 µ/ml
En EE. UU. se ha adoptado la estrategia basada en la determinación de T4, mientras que en Europa y Japón se investiga la TSH en primer lugar. La estrategia de la T4 es más costosa desde el punto de vista económico, pero permite la detección de HC secundarios, terciarios y por déficit de globulina transportadora de tiroxina (TBG) sin elevación de la TSH, si bien en este caso serán necesarios para el diagnóstico los test de TSH y TRF, y determinaciones de TBG, estos trastornos son mucho más raros, de menor gravedad.
Algunos niños prematuros, así como los recién nacidos que han precisado cuidados intensivos o han recibido tratamiento con dopamina o yodo en sus diversas formas de administración, tienen elevaciones de la TSH y amplias fluctuaciones en los niveles de T3 y T4 que impiden valorar correctamente los niveles de TSH. Los programas que se basan en dos determinaciones informan que hasta un 10% de los casos de HC se diagnostican con la segunda determinación8. Según la American Academy of Pediatrics, la prevalencia de HC para la segunda determinación es de 1:30 000 y suelen ser niños prematuros y de muy bajo peso al nacer. Es conocido que algunas situaciones, como la prematuridad, las cardiopatías o la administración de fármacos como dopamina, amiodarona o los contrastes yodados, pueden causar elevaciones de la TSH. Está indicado por tanto hacer una segunda determinación en prematuros menores de 32 semanas o de peso inferior a 1500 g. Algunas comunidades establecen una segunda determinación de rutina.
Casi todos los autores proponen que el cribado la primera determinación se realice a las 48 horas de vida2,4-7.
Fenilcetonuria
La fenilcetonuria es un error innato del metabolismo en el que existe un defecto de hidroxilación de la fenilalanina (FA), que no puede convertirse en tirosina como consecuencia del déficit de fenilalanin-hidroxilasa (FAOH) o de la dihidropterina reductasa (DPHR). Esta última enzima necesita para su actuación de la presencia de tetrahidrobiopterina (BH4). La tirosina se convierte así en un aminoácido esencial para el organismo, a la vez que se produce un aumento de fenilalanina en sangre y aumenta su transaminación como vía metabólica alternativa. Se acumulan, asimismo, los ácidos fenil-pirúvico, feniláctico y fenilacético. El defecto en la síntesis de FAOH se debe a una anomalía génica localizada en el cromosoma 12, y el de la DPHR en el cromosoma 4. Existen también formas con déficits parciales. En conjunto, las hiperfenilalaninemias tienen una prevalencia en España de 1:9201. Las formas más graves 1:19 747 y las moderadas 1:12 4222.
La FC produce un retraso psicomotor y un deterioro intelectual irreversibles en poco tiempo. Estos trastornos pueden prevenirse si se instaura precozmente una dieta pobre en fenilalanina. Los niños con FC suelen ser rubios, de tez pálida y con un olor característico a paja mojada. El cribado de FC es muy eficaz, ya que antes del cribado el 85% de los niños con FC tenía un cociente intelectual (CI) menor de 40, y un 37%, menor de 10. Actualmente, con el cribado, cerca del 95% de los niños con FC tiene un CI normal. Los elevados niveles de FA en sangre dan lugar a alteraciones estructurales del sistema nervioso central, con interferencia en el proceso de maduración cerebral. Estas alteraciones neuropatológicas producen un grave retraso mental si no se inicia precozmente una alimentación pobre en FA.
El cribado de la FC se realiza mediante fluorometría, cromatografía en papel o en capa fina y últimamente mediante EMT2,4-6. La clásica prueba de Guthrie ha quedado obsoleta. Para el cribado se considera adecuada la toma de muestras entre las 24 horas y los siete días de vida (no antes de las 12 horas). Los puntos de corte de FA son de 150 µM (2,5 mg/dl) y el punto de corte para cociente fenilalanina/tirosina es de 2,5. Para una determinación correcta es necesario asegurarse de que se haya realizado una ingesta adecuada de proteínas y que no esté recibiendo nutrición parenteral.
Es preciso instaurar precozmente un régimen dietético. Los niveles de FA elevados, aun durante cortos períodos de tiempo, pueden producir serios trastornos de conducta y eccema facial. Se deben monitorizar los niveles de FA, manteniéndolos entre tres y seis mg/dl. Hay controversia sobre el tiempo que debe durar este régimen alimentario. Existen, sin embargo, recomendaciones en las que se insiste en prolongarlo de forma indefinida.
En los pacientes que responden al cofactor se administra BH4.
Hiperplasia suprarrenal congénita
La hiperplasia suprarrenal congénita (HSC) se debe generalmente a la ausencia o disminución de la enzima 21-hiidroxilasa, que da lugar a un bloqueo en la síntesis de cortisol, con aumento secundario en la síntesis de andrógenos y virilización del feto. Con gran frecuencia, se produce un cuadro de pérdida salina hacia las dos semanas de vida. La prevalencia en España es de alrededor de 1:12 718 en las CCAA en las que se realiza5.
El tratamiento con hidroxicortisona estabiliza el problema y permite un crecimiento normal. En algunos casos se precisa añadir mineralcorticoides. El cribado y la detección de portadores son posibles mediante la determinación de la 17-OH-progesterona por inmunoensayo, fluorometría o espectrometría de masas en tándem. El punto de corte par la determinación de 17-OH-progesterona es 10-30 ng/ml, y para recién nacidos de bajo peso y prematuros, inferior a 50 ng/ml. El cribado neonatal con 17-OH-progesterona tiene algunos problemas, con tasas de falsos positivos por encima del 1%. Esto se debe a que pueden darse reacciones cruzadas con otros esteroides que están elevados por estrés y también por la prematuridad. Algunos centros de cribado proponen una estrategia con dos muestras que mejore la especificidad. Otra alternativa es la detección simultánea de 17-OH-progesterona, cortisol y androstendiona mediante EMT.
Fibrosis quística de páncreas
La fibrosis quística de páncreas (FQP) se hereda con carácter autosómico recesivo. Se produce por una anomalía en el gen CFTR en el locus 7q31. Se produce por una mutación del gen que codifica la proteína reguladora de la conductancia transmembrana.
Su frecuencia en España es 1:3449. Se caracteriza por una anomalía exocrina generalizada, con una anormal viscosidad de todas las secreciones exocrinas que ven de esta forma dificultada su eliminación, acumulándose en los conductos excretores. En el aparato respiratorio conduce a bronconeumopatía crónica, sinusitis y poliposis nasal. En el tracto digestivo se produce en muchos casos un íleo meconial perinatal, pero también una insuficiencia pancreática con esteatorrea y desnutrición, alteraciones hepáticas con fibrosis y finalmente una verdadera cirrosis biliar. En el aparato reproductor masculino da lugar a azoospermia o agenesia de vasos deferentes. La anomalía en el gen CFTR lleva, asimismo, a una alteración en los canales del cloro, con una mayor concentración de cloruro de sodio en el sudor. En situaciones de sudoración profusa se pueden llegar a producir cuadros de deshidratación hiponatrémica. Es típica la colonización precoz porPseudomonas aeruginosa yBurkholderia cepacia. La enfermedad es de penetrancia variable. Desde que es posible analizar el ADN genómico, se conocen más de 1000 mutaciones para el gen CFTR. La más frecuente es la delta F508 (DF508), que en España representa el 50% de todas las mutaciones y el 70% en otros países.
Técnica de cribado
El primer intento de cribado para la FQP se produjo en los años 50 mediante el test del meconio. Desde finales de los 70 se determina la tripsina inmunorreactiva (TIR) en mancha de sangre seca, ya que la concentración plasmática de esta sustancia está aumentada en la FQP. Se utiliza la inmunofluorescencia de tiempo retardado y el enzimoinmunoanálisis. Pueden considerarse normales cifras entre 90 y 200 ng/ml. Su confirmación requiere de una segunda determinación y un test del sudor, pasado un mes. Posteriormente se ha desarrollado el análisis del ADN. Actualmente, el cribado se realiza con una técnica combinada. Se utiliza la misma mancha de sangre que para otras metabolopatías. Se determina la TIR en primera instancia y, si esta supera un punto de corte de 110 ng/ml, se realiza sobre la misma muestra un análisis del ADN, buscando la mutación DF508 o bien otras mutaciones. Con esta técnica secuencial TIR/ADN, la sensibilidad es prácticamente del 100% y la especificidad del 99,5%.
El cribado de la FQP mejora los resultados de la enfermedad, y las poblaciones sometidas a cribado evolucionan mejor que las que se abandonan a la suerte del diagnóstico clínico2-6. El cribado permite anticipar y prevenir la deshidratación hiponatrémica por situaciones de aumento de la sudoración. El crecimiento y desarrollo de las poblaciones de FQP sometidas a cribado es mejor durante los primeros diez años de vida, ajustando los datos para un mismo nivel de función pancreática y también es menor el deterioro de la función pulmonar. El cribado permite, asimismo, la prevención o el retraso de la colonización porPseudomonas, en muchos casos determinante de la evolución respiratoria de los niños con FQP. Algunos estudios muestran un menor número de infecciones respiratorias a los dos años de vida en niños con FQP sometidos a cribado, respecto a los no sometidos a cribado.
Drepanocitosis o anemia de células falciformes
La incidencia de anemia de células falciformes (ACF) en el colectivo subsahariano es de alrededor del 1,18%; en los norteafricanos, del 0,05%, y en los afrocaribeños, del 0,2%. La ACF se debe a una anomalía de la hemoglobina (HbS). La HbS es una variante de la HbA de los adultos2.
La enfermedad cursa con anemia crónica moderada o grave y crisis vasooclusivas que cursan con síntomas dolorosos en cualquier punto del organismo y posibilidad de sepsis, sobre todo por Streptococcus pneumoniae, Haemophilus influenzae y Neisseria meningitidis por asplenia funcional. En la ACF está indicada la profilaxis con penicilina y la vacunación antineumocócica y antimeningocócica para prevenir complicaciones infecciosas. Existen diversas formas de presentación de la ACF según el número de alelos afectados o la presencia del gen de la ß-talasemia. Por ello, la forma de presentación va desde el rasgo de ACF hasta formas variables de anemia y crisis vasooclusivas.
Técnica de cribado
El cribado se realiza mediante detección de HbS y HbA, F, A2, S, C, E y D mediante cromatografía líquida de alta resolución de intercambio catiónico, electroforesis capilar, isoelectroenfoque y fluoroinmunoanálisis Otras técnicas más sofisticadas serían las técnicas de análisis genético. El cribado de la ACF estaría indicado en población de origen norteafricana, subsahariana, afroamericana y del sudeste asiático, dada la alta prevalencia en estos colectivos. Debido al elevado porcentaje de población de origen subsahariano que vive actualmente en España, es conveniente que en un futuro se plantee el cribado neonatal universal de este trastorno en España, es por tanto muy oportuno que en el mencionado acuerdo del Consejo Interterritorial se haya planteado el cribado neonatal universal de este trastorno2-6.
Deficiencia de acil CoA deshidrogenasa de cadena media (MCAD)
Es un trastorno por defecto en el gen MCAD en el locus I p31.Se produce un defecto de la betaoxidación de los ácidos grasos de cadena media. La incidencia está entre 1:3000-1:17 000. Se dispone de los datos de Galicia (1:11 333). La herencia es autosómica recesiva. La clínica es de vómitos, estupor, letargia que puede progresar a coma y fracaso cardiorrespiratorio, convulsiones y muerte. Puede haber hepatomegalia y pruebas hepáticas, amonio y pruebas de coagulación alteradas. Suele haber hipoglucemia durante los episodios de agudización con cetonas bajas, aunque en ocasiones la cetonuria puede llegar a ser normal, por lo que una hipoglucemia con cetonas normales no puede excluir el diagnóstico de MCAD2-6.
El cribado se realiza mediante la determinación de acilcarnitinas en sangre con elevaciones de la octanoilcarnitina (C8) y de la decnoilcarnitina (C10) y del cociente C8/C10; la carnitina total suele estar disminuida y la esterificada suele aumentar.
El cribado se realiza mediante EMT.El tratamiento consiste en evitar los periodos prolongados de ayuno, suplementar con carnitina y evitar la ingesta de ácidos grasos de cadena media. Durante los periodos de descompensación, aporte de hidratos de carbono orales o si fuera el caso de glucosa intravenosa.
Acidemia glutárica tipo I (AGI)
Es un defecto en el gen GCDH en el locus I 9p13.2 por deficiencia de glutaril CoA deshidrogenasa. La prevalencia es de 1:75 000 y en Galicia de 1:34 000. Es de herencia autosómica recesiva. La clínica es de afectación neurológica tras sufrir una crisis encefalopática tras fiebre, vacunación o intervenciones quirúrgicas. Se produce un daño de los núcleos estriados con cuadros de distonía, ataxia y espasticidad. Hay dos perfiles bioquímicos diferentes. La de los altos excretores de ácido glutárico (>100 mmol/mol de creatinina en orina) y la de los bajos excretores (<100 mmol/mol creatinina)2-6.
El cribado se realiza mediante EMT por determinación de la glutarilcarnitina (C5-DC) y del cociente C5-DC/C16.
El tratamiento implica suplementar con carnitina y riboflavina y la restricción proteica con aporte libre de aminoácidos precursores.
Acidemia isovalérica (AIS)
Es un déficit de isovaleril deshidrogenasa localizado en el gen IVD locus I 5p I 4-15.
Se trata de un trastorno del catabolismo de la leucina. La prevalencia varía entre 1:62 500 en Alemania y 1:250 000 en EE. UU. Es autosómica recesiva. La forma de presentación adopta dos formas: la precoz neonatal, más grave, que se presenta en la primera semana con vómitos, rechazo de las tomas, estupor y coma2-6.
La forma crónica, que suele aparecer a partir del año de vida también en forma de crisis con estos síntomas, si bien no tan graves y desencadenados por ingesta proteica o infecciones. En algunos casos, no en todos, se produce un deterioro del desarrollo psicomotor. Es característico el olor a pies sudados.
El cribado se realiza mediante EMT por detección de acilcarnitinas, fundamentamente C5 (isovalerilcarnitina). La AIS no puede diferenciarse mediante EMT de la deficiencia de 2-metilbutiril CoA deshidrogenasa, por lo que es necesario realizar otras pruebas.
El tratamiento consiste en evitar periodos prolongados de ayuno, restricción de leucina y suplementos de carnitina2-6.
ESPECTROMETRÍA DE MASAS EN TÁNDEM
La EMT es una tecnología relativamente nueva dentro de la bioquímica analítica, mediante la cual se identifican compuestos químicos ionizados sometidos a un campo magnético. El aparato de EMT dispone de dos cámaras. En la primera se ioniza el compuesto químico mediante bombardeo de partículas que lo fragmenta en moléculas y una segunda cámara donde actúa un potente campo magnético. La velocidad de desplazamiento de cada molécula dentro del campo magnético nos permite identificarlas de forma muy específica5,6.
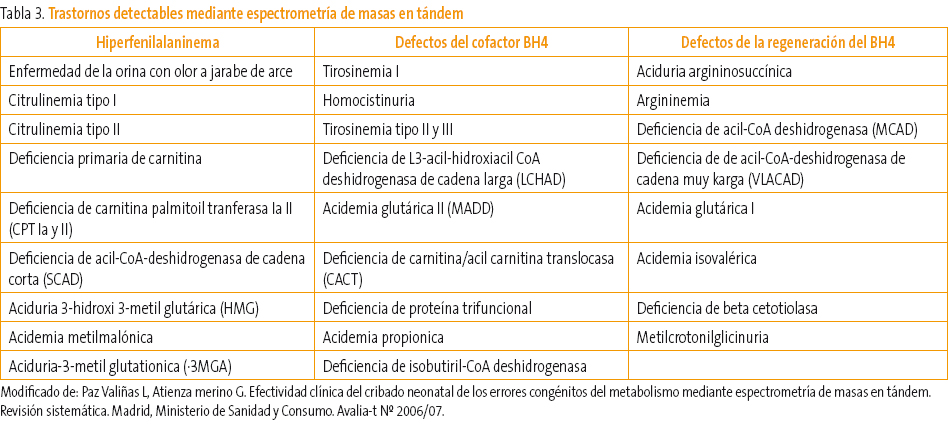
En la Tabla 3 se muestran los trastornos que pueden ser identificados mediante EMT.
Tabla 3. Mostrar/ocultar
TÉCNICA DE RECOGIDA DE LA MUESTRA
Se toma una misma muestra para el cribado de HC, FC, HSC y FQP. En primer lugar se desinfecta el talón con alcohol de 70° y se seca posteriormente. Se punciona con una lanceta estéril y desechable en una de las caras laterales de la parte plantar del talón. Se deja que se forme espontáneamente la primera gota de sangre, que se retira con una gasa estéril. Después se coloca el papel de filtro homologado en contacto con la segunda gota de sangre, hasta que empapa toda la superficie destinada a la mancha de sangre. La mancha debe rellenar todo el círculo dibujado en el papel y empapar bien por ambos lados, de modo que la mancha sea igual por el anverso que por el reverso. La sangre debe recogerse de una sola vez. Los papeles se secan al aire durante tres horas en posición horizontal, sin colocar nada encima. Deben conservarse en lugar seco y protegidos de la luz. Finalmente pueden enviarse por correo al laboratorio de referencia.
MANEJO DURANTE LA PUNCIÓN DEL TALÓN
Los cambios fisiológicos provocados por el dolor pueden producir desorganización en el desarrollo de las conexiones neuronales. Existen ya numerosos estudios que demuestran que medidas no farmacológicas simples como el amamantamiento y el contacto piel con piel disminuyen el dolor a través de mecanismos relacionados con la liberación de endorfinas. Otras medidas no farmacológicas son la administración de soluciones de sacarosa o la estimulación táctil. La punción del talón es dolorosa y debería realizarse mientras el lactante se amamanta o, si no es posible, mediante alguna de las medidas mencionadas7-9.
INFORMACIÓN A LOS PADRES Y CONSENTIMIENTO INFORMADO
Los padres deben ser informados de cuantos procedimientos se realizan y de la finalidad de los mismos. En el caso del cribado neonatal, la información es compleja, por lo que pueden servir de ayuda los materiales elaborados con este propósito10,11. El consentimiento por escrito no es imprescindible pero es necesario cerciorarse de que entienden la finalidad del programa y de que prestan su conformidad.
BIBLIOGRAFÍA
- Soriano Faura J. Estudio descriptivo sobre actividades preventivas por CCAA según la información aportada por las juntas ejecutivas de las asociaciones federadas de la Asociación Española de Pediatría de Atención Primaria. En AEPap ed. Curso Actualización Pediatría 2012. Madrid.
- Marín Soria JL, Aldamiz-Echevarría L, Castiñeiras Ramos DE, Dalmau Serra J, Fernández Sánchez A, González Lamuño D, et al. Programas de cribado neonatal en España: actualización y propuesta de futuro. Madrid: Ministerio de Sanidad y Consumo; 2009.
- Andradas Aragonés E, Lizarbe Alonso VM, Labrador Cañadas MV, Alfaro Allona C, Díaz Torres P, Izquiedo Martínez M. Resumen ejecutivo del grupo de expertos sobre concrecion de cartera de servicios para cribado neonatal. Madrid. Ministerio de Sanidad, Servicios Sociales e Igualdad; 2013.
- Thomason M, Lord J, Murray D, Chalmers RA, Littlejohns P, Addison M, et al. A Systematic re-view of evidence for the appropriateness of neo-natal screening programmes for inborn errors of metabolism. J Public Health. 1998;20:331-43.
- Paz Valiñas L, Atienza merino G. Efectividad clínica del cribado neonatal de los errores congénitos del metabolismo mediante espectrometría de masas en tándem. Revisión sistemática. Madrid, Ministerio de Sanidad y Consumo. Avalia-t Nº 2006/07.
- Pandor A, Eastham J, Beverley J, Chilcott S. Clinical effectiveness and cost-effectiveness of neo-natal screening for inborn errors of metabolism using tandem mass spectrometry: a systematic review. Health Technol Asses. 2004;8812:1-312.
- Rodríguez Sánchez MD, Pandilla Esteban ML, González Sicilia IG, Lorenzo Navarro L, Bittini Copano A, Dulín Iñíguez E, et al. Hipotiroidismo congénito. Detección precoz. Diagnóstico y tratamiento. Acta Pediatr Esp. 1994;52:217-27.
- Codipietro L, Ceccarelli M, Ponzone A. Breastfeeding or oral sucrose solution in term neonates receiving heel lance: a randomized, controlled trial. Pediatrics. 2008;122;e716-21.
- Yamada SB, Ohlsson A. Sucrosa para la analgesia en recién nacidos sometidos a procedi-mientos dolorosos (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, 2008 Número 4. Oxford: Update Software Ltd. Disponible en http://www.update-software.com (Traducida de The Cochrane Library, 2008 Issue 3). Chichester, UK: John Wiley & Sons, Ltd.
- Asociación Española de Cribado Neonatal [en línea] [actualizado el 14/12/2008; consultado el 09/08/2009]. Disponible en http://aecne.es/pdf/datos2007.pdf
- Galbe J y Grupo Previnfad ¿En qué consiste la prueba del talón? En: Familia y Salud. Aepap [en línea] [consultado el 6/6/2013]. Disponible en http://www.familiaysalud.es/podemos-prevenir/cribados-neonatales/en-que-consiste-la-prueba-del-talon