

Lactante de 15 meses con broncoespasmos de repetición
2 Pediatra. Tutora MIR de Pediatría. CS Puerto de la Torre. Málaga (España).
3 Pediatra. Tutora de MIR de pediatría. CS Delicias. Málaga (España).
RESUMEN: PUNTOS CLAVE PARA EL PEDIATRA DE ATENCIÓN PRIMARIA
Lactante de 15 meses con broncoespasmos de repetición
SUMMARY: KEY POINTS FOR PRIMARY CARE PEDIATRICIANS
PUNTOS CLAVE
- Un niño con broncoespasmos repetidos y mala respuesta al tratamiento con broncodilatadores debe hacernos sospechar una patología de base.
- La fibrosis quística es un fenotipo clínico que puede o no presentar la mutación característica, por lo que hay que estar atentos a pesar de tener un screening negativo.
- La fibrosis quística es la enfermedad hereditaria grave más frecuente de la raza blanca, donde la morbilidad y la mortalidad vienen condicionados especialmente por la afectación pulmonar.
- El diagnóstico temprano mejora el pronóstico y la supervivencia; un 30% de los niños son diagnosticados tardíamente debido a las diferencias autonómicas de cribado neonatal.
- Las enfermedades respiratorias son la manifestación más frecuente de la enfermedad.
- Un niño sano con pruebas positivas no es un enfermo, pero requiere seguimiento.
- Mantener el estado nutricional adecuado es el pilar fundamental del tratamiento, por lo que es muy importante la educación nutricional de los padres desde el diagnóstico.
CASO CLÍNICO
Niña de un año de edad que acudió a consulta por mala ingesta alimentaria habitual, en cantidad y calidad, tos intensa, escasa mucosidad blanquecina y cierta dificultad respiratoria. En las últimas tres semanas había iniciado un cuadro de deposiciones diarreicas en número de tres-cuatro al día sin productos patológicos. Desde los cuatro meses de vida presentaba broncoespasmos de repetición desencadenados por infecciones virales de difícil control con broncodilatadores y corticoides inhalados. Dada la mala respuesta al tratamiento se decide ampliar el estudio.
Antecedentes
- Antecedentes clínicos: bronquiolitis, hiperreactividad bronquial, dermatitis atópica y retraso ponderoestatural.
- Antecedentes familiares: hermano varón asmático.
Exploración física
Buen estado general. Bien hidratada y perfundida. Escaso panículo adiposo. Peso 7,5 kg. (percentil 2 [p2]), talla 78 cm (p92). No presentaba taquipnea, distrés ni tiraje. En la auscultación pulmonar destacaba una discreta hipoventilación en la base pulmonar izquierda y crepitantes en el campo pulmonar derecho. La faringe estaba hiperémica con mucosidad abundante en el cavum. El abdomen era blando y depresible, sin masas ni megalias y con dolor difuso a la palpación.
Pruebas complementarias
Pruebas realizadas en el centro de salud
- Hemograma, perfil lipídico y hepático, proteínas totales, albúmina, proteína C reactiva, glucemia, creatinina e iones dentro de los valores normales.
- Análisis de heces de 24 horas mediante espectroscopia de reflectancia en infrarrojo cercano: Grasas 27,4 g (valor normal [VN] <3 g), nitrógeno 5,3 g (VN <0,65 g).
- Radiografía de tórax: engrosamiento peribronquial parahiliar bilateral, con algunas atelectasias laminares por tapones de moco. Sin condensaciones.
- Test del sudor positivo. Conductividad 115 mEq/l.
Pruebas realizadas en el hospital
- Genética: mutación F508del en homocigosis.
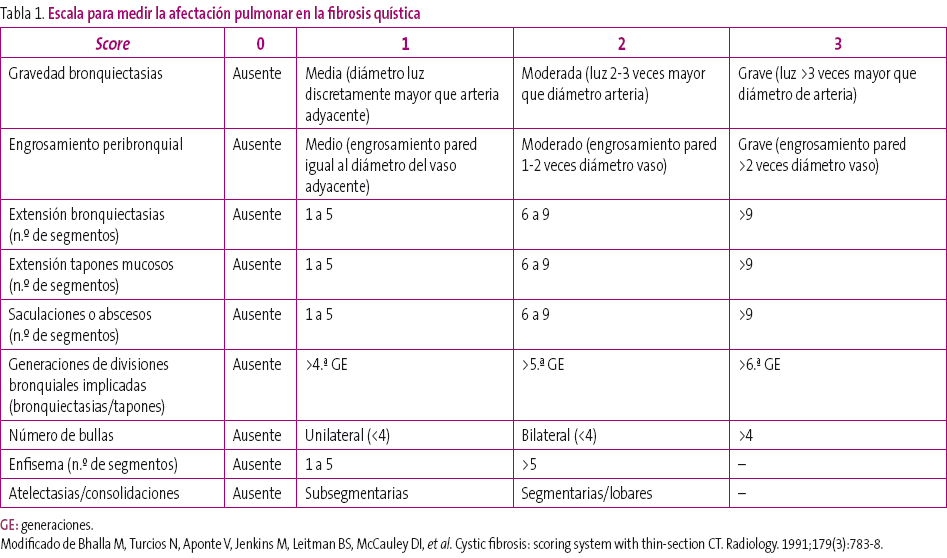
- Tomografía computarizada torácica: Bhalla 24 (Bhalla es la escala para medir la afectación pulmonar en fibrosis quística. La puntuación consiste en restarle a 25 los puntos de las lesiones presentes en la TC, siendo por tanto más grave aquellas con un Bhalla menor) (Tabla 1)1.
- Inmunoglobulinas normales.
- Prueba de radioalergosorbencia (RAST): negativa para Aspergillus.
- Cultivo de exudado faringoamigdalar: flora normal.
Tabla 1. Mostrar/ocultar
Evolución
Las pruebas realizadas confirmaron el diagnóstico de fibrosis quística asociada a insuficiencia pancreática y hepatopatía leve. Tras iniciar tratamiento con ácido ursodeoxicólico, suplementación alimenticia, profilaxis antibiótica y fisioterapia respiratoria, la paciente presentó una mejoría importante, aumentando la velocidad de crecimiento a 1 cm/mes. Desde el diagnóstico, la paciente se ha colonizado por Pseudomonas aeruginosa en dos ocasiones, con buena respuesta al tratamiento.
DISCUSIÓN
La causa más frecuente de broncoespasmos en el lactante son los virus (virus respiratorio sincitial [VRS] y rinovirus). Sin embargo, en ocasiones pueden deberse a otras enfermedades con las que debemos plantear el diagnóstico diferencial.
La fibrosis quística es un fenotipo clínico caracterizado por uno o más de los siguientes signos clínicos: inflamación respiratoria crónica, infección por organismos típicos (tales como Staphylococcus aureus o Pseudomonas aeruginosa), insuficiencia pancreática, sinusitis grave, pólipos nasales, cirrosis biliar y electrolitos de sudor aumentados. Se corresponde en la gran mayoría de los casos con la mutación genética autosómica recesiva del brazo largo del cromosoma 7, que codifica la proteína reguladora del intercambio de cloro, controlado por AMPc en las membranas (cystic fibrosis transmembrane conductance regulator [CFTR])2. La imposibilidad para realizar el transporte de cloro conlleva una secreción inadecuada de líquido, una inadecuada hidratación de las macromoléculas y la alteración de las propiedades fisicoquímicas de las secreciones de los órganos afectos, que se espesan obstruyendo sus conductos; no es, sin embargo, esta su única etiología, ya que en una minoría de pacientes se ha comprobado que existe una hiperabsorción de sodio activa asociada a la absorción de agua y deshidratación en la superficie de la vía aérea, presentando CFTR normal.
Manifestaciones clínicas
La presentación de la fibrosis quística varía en cada paciente y debemos estar atentos en la consulta de Atención Primaria para captar los síntomas de alarma.
Algunas complicaciones frecuentes de esta enfermedad, por su fisiopatogenia, son el reflujo gastroesofágico, la bronquiolitis positiva para VRS, las atelectasias, la aspergilosis, el cor pulmonale, el neumotórax, el neumomediastino, la insuficiencia respiratoria y la hemoptisis por infección pulmonar crónica.
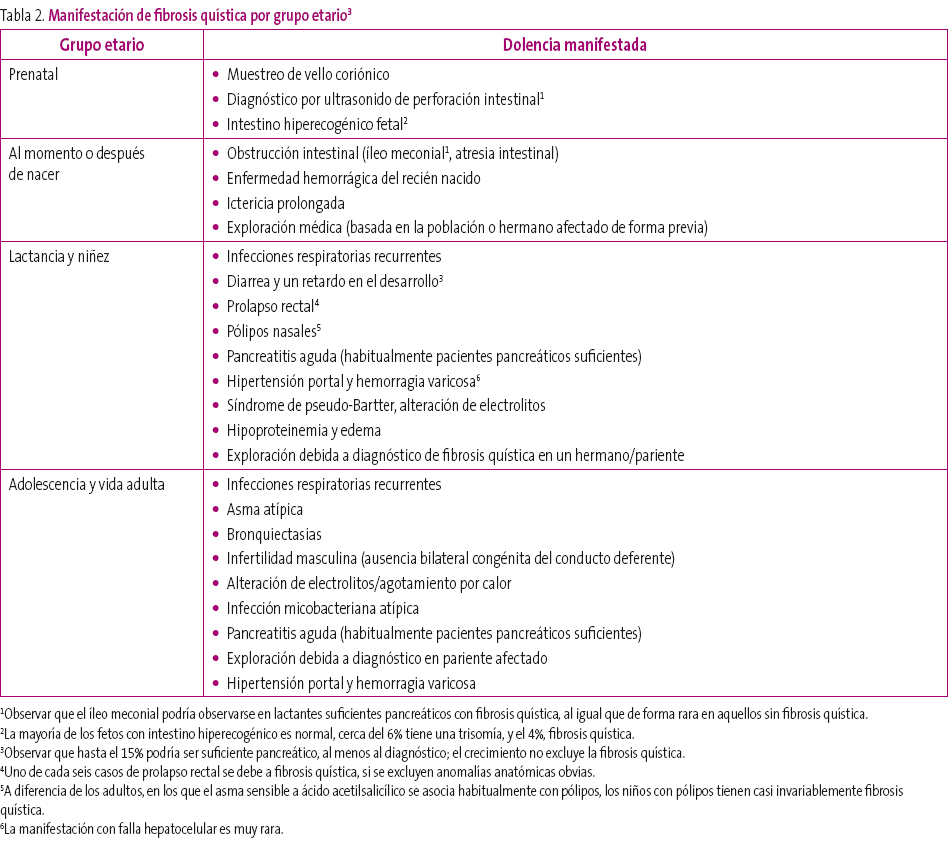
En la Tabla 2 se exponen las manifestaciones clínicas más frecuentes en cada tramo de edad.
Tabla 23. Mostrar/ocultar
Diagnóstico
Cribado neonatal
Debido a que la tripsina se eleva en sangre en neonatos con fibrosis quística, se ha establecido como método de cribado los niveles de tripsina inmunorreactiva (TIR) en la prueba del talón a los 3-5 días de vida4. Aquellos que resulten positivos (TIR > 60) deben repetirse a los 25-40 días de vida y, en caso de resultar positivos de nuevo (TIR > 40), deben confirmarse mediante el test del sudor. Si este resulta positivo, se procederá al estudio de las mutaciones genéticas más frecuentes. Los falsos positivos de esta determinación pueden deberse a la raza, el estado de portador o el nivel de salud perinatal. En cuanto a los falsos negativos, el 1-5% se ve influido por la edad de realización de la prueba y por la presencia de íleo meconial.
Este cribado esta disponible mediante recogida de sangre en papel secante en Madrid, Castilla y León, Murcia, Aragón, País Vasco, Cataluña, Baleares, La Rioja y Andalucía, en la maternidad, y en Canarias, Galicia y Extremadura en Atención Primaria en los primeros 3-5 días de vida5.
Mide la concentración de cloruro en el sudor. Se precisan dos determinaciones positivas y descartar otras patologías que puedan positivizar la prueba, tales como insuficiencia suprarrenal, anorexia nerviosa, hipoparatiroidismo, hipotiroidismo, diabetes insípida nefrogénica y síndrome nefrótico.
Se establece el diagnóstico de fibrosis quística clásica en presencia de al menos una característica fenotípica (enfermedad sinopulmonar crónica, alteraciones digestivas y nutricionales, síndromes de pérdida de sal, ausencia bilateral de conductos deferentes), junto con una concentración de cloro en sudor >60 mmol/l. En estos pacientes, generalmente se detectan dos mutaciones causantes de enfermedad en el gen CFTR, pueden tener o no insuficiencia pancreática y su evolución clínica es variable. Se establece el diagnóstico de fibrosis quística no clásica o atípica si se halla al menos una de las características fenotípicas y la prueba del sudor con resultado borderline (cloro 30-60 mmol/l) junto con la detección de dos mutaciones y/o una diferencia de potencial nasal (DPN) alterado. Estos pacientes suelen tener suficiencia pancreática y enfermedad pulmonar más leve que los que sufren una clásica, y la afección clínica puede ser de uno o varios órganos8.
Diferencia del potencial nasal transepitelial
En fibrosis quística será más negativa: la composición electrolítica del líquido periepitelial viene determinada por la capacidad de las células epiteliales de transportar iones, como el cloro y el sodio, generando una diferencia de potencial transepitelial que puede medirse in vivo en la mucosa nasal4. La medición de este potencial permite establecer un patrón de anormalidad en pacientes con fibrosis quística, como consecuencia del aumento en la reabsorción de sodio que lo hace más electronegativo (media de DPN en la FQ-46mv frente a DPN en la población sana-19mv).
Estudio genético
Para el diagnóstico se requiere la demostración de la existencia de una mutación causante de alguno de los mecanismos básicos que alteran la función de la CRTF en ambos alelos del cromosoma 7 (dos mutaciones en total)9. La existencia de una sola mutación los convierte en portadores pero no en enfermos. Las clases de mutaciones son:
- Clase I. No hay síntesis de proteína: G542X.
- Clase II. Bloqueo de la correcta traducción y alteración de su localización en el aparato de Golgi, como resultado es retenida en el retículo endoplásmico y eventualmente degradada por los mecanismos de control de calidad: ΔF508, N1303K.
- Clase III. Proteínas mutadas incapaces de abrirse al estímulo del AMPc: G551D.
- Clase IV. Afecta a la conductancia del cloro, no lo reconoce: R117H, R334W.
- Clase V. Reducidos niveles de proteína: A455E.
Las mutaciones I, II y III están asociadas a insuficiencia pancreática, en la IV tiene reducido el flujo a través del canal; pero hay suficiente actividad residual como para dar un fenotipo de suficiencia pancreática.
Las mutaciones AF508 y G542X son las que se han detectado con mayor frecuencia en nuestro medio en un 58%.
En caso de pruebas positivas y niño sin síntomas no se considera afecto de la enfermedad, pero implica la necesidad de un seguimiento.
Seguimiento y evolución
- Exploración clínica: se debe comprobar el incremento estaturoponderal y examinar los síntomas respiratorios y/o la presencia de acropaquias.
- Pruebas de imagen torácicas: radiografía al diagnóstico y TC anual. La frecuencia de realización dependerá de la clínica y los hallazgos.
- Determinación de elastasa fecal en visitas a Gastroenterología para establecer insuficiencia pancreática en caso de tenerla (insuficiencia pancreática = elastasa <100).
- Espirometría en cada visita al Servicio de Neumología (aproximadamente cada tres meses) por el deterioro funcional respiratorio que conlleva (el volumen espiratorio forzado en un segundo [FEV1] es el mayor predictor pronóstico).
- Cultivo de esputo o exudado faringoamigdalar en caso de no tener capacidad para expectorar cada 1-2 meses y en infecciones agudas.7
Tratamiento
El tratamiento de los pacientes con fibrosis quística debe ser multidisciplinario, incluye al neumólogo, al gastroenterólogo, al fisioterapeuta, al nutricionista, al psicólogo y al pediatra de Atención Primaria, todos profesionales importantes en su seguimiento6.
Tratamiento respiratorio
Engloba las siguientes terapias:
- Antibióticos: rara vez se conseguirá erradicar los gérmenes, por lo que su función será controlar la infección y no curar. El tratamiento a elegir dependerá del germen y su sensibilidad. (P. Aeruginosa: cefalosporina de tercera generación y aminoglucósido. En caso de resistencia a cefalosporina se usarán carbapenemas). En las exacerbaciones moderadas y graves se usará administración intravenosa en altas dosis durante 14-21 días, no es necesario el ingreso hospitalario. En las exacerbaciones leve-moderadas se usará la vía oral (ciprofloxacino, trimetropim-sulfametoxazol y cloramfenicol) e inhalada (colimicina, tobramicina), siendo esta última la vía de tratamiento de mantenimiento. La profilaxis de S. aureus es controvertida hoy día.
- Broncodilatadores: se utilizan antes de la fisioterapia y para el control de secreciones.
- Mucolíticos: la DN-asa reduce la viscoelasticidad del esputo y el suero salino hipertónico al 7% inhalado aumenta el líquido pericelular. Las terapias mucolíticas tradicionales como la acetilcisteína no son útiles.
- La amilorida inhalada cuatro veces al día antagoniza los canales de sodio y disminuye el declive de la función pulmonar.
- Antiinflamatorios, para contrarrestar la inflamación por la persistente infección bacteriana: la azitromicina, las estatinas y el metotrexato reducen las exacerbaciones respiratorias. También se usa el ibuprofeno. Los corticoides orales son útiles pero presentan muchos efectos secundarios.
- Fisioterapia respiratoria.
Tratamiento digestivo
Engloba las siguientes terapias:
- Suplementación enzimática pancreática: 500-2500 U lipasa/kg/comida (con aperitivos y snacks utilizar la mitad de la dosis). Nunca se debe sobrepasar las 10 000 U lipasa/kg/día o las 4000 U lipasa/g de ingesta grasa. En caso de no controlarse la esteatorrea, se intentará disminuir la acidez gástrica o aumentar la alcalinización duodenal.
- Vitaminas liposolubles: vitamina A 5000-10 000 U/día, D 400-800 U/día y E 50/200 U/día. La vitamina K será necesaria en caso de infecciones y antibióticos frecuentes o colestasis.
- Ácido ursodeoxicólico para fluidificar bilis en el caso de existir enfermedad hepática.
- Lavados con polietilenglicol en caso de obstrucción intestinal.
- Aporte calórico elevado (200% del recomendado en niños sanos: 20% proteínas, 50% hidratos de carbono y 30% grasas). Deben evitarse las restricciones de grasas.
- Suplementos nutricionales: se recomiendan en pacientes con infecciones recurrentes, insuficiencia respiratoria, percentiles <15 o disminución de la velocidad de crecimiento.
Terapia génica
En la actualidad se está investigando la modificación del curso de la enfermedad a través de correctores de las mutaciones: genisteína, aminoácido que reduce el bloqueo de transporte del cloro por el CFTR, modificadores del transporte iónico, y corrección del CFTR por clonación de su ADN complementario e introducción del mismo mediante liposomas o adenovirus en el organismo. Hasta el momento, los resultados obtenidos muestran correcciones transitorias de la mutación.
BIBLIOGRAFÍA
- Bhalla M, Turcios N, Aponte V, Jenkins M, Leitman BS, McCauley DI, et al. Cystic fibrosis: scoring system with thin-section CT. Radiology. 1991;179(3):783-8.
- Dickinson F, Batlle MC, Razón Behar R, Ramos Carpenter L, Pérez Monrás M. Caracterización epidemiológica de pacientes pediátricos con fibrosis quística. Rev Cubana Pediatr. 2005;77(2).
- Asensio de la Cruz O, Bosque García M, de los Ríos Pérez A. Fibrosis quística y sus manifestaciones respiratorias. Pediatr Integral. 2012;16(2):156-69.
- Martínez Calvo AV, Paz-Valiñas L, García-Vega FJ. Cribado neonatal de la fibrosis quística. Santiago de Compostela: Servicio Galego de Saúde, Axencia de Avaliación de Tecnoloxías Sanitarias de Galicia, avalia-t; 2004 [en línea] [consultado el 10/12/2012]. Disponible en: http://www.sergas.es/cas/Servicios/docs/AvaliacionTecnoloxias/Fibr%20quistica%20%20INF2004%2002.pdf
- Metabolopatías. Qué cribados neonatales se hacen en según qué comunidad autónoma española (encuesta 2010). El GIPI [en línea]. Disponible en: https://spreadsheets.google.com/pub?key=0AsIJdw22PvEwdHQ4NUlZbjlqZThPdDBtMlQyYnI3LWc&output=html
- Escobar Castro H, Sojo Aguirre A, Gil Ortega D, Nadal Ortega JM. Fibrosis quística. Protocolos diagnóstico-terapéuticos de gastroenterología, hepatología y nutrición pediátrica. Madrid: SEGHNP-AEP; 2010 [en línea] [acceso 10/12/2012]. Disponible en: http://www.gastroinf.com/Protocolos%20SEGHNP
- Bush A. Diagnóstico de fibrosis quística: lo fácil, lo difícil, lo imposible. Neumol Pediatr. 2010;5:15-22.
- Barrio Gómez de Agüero MI, García Hernández G, Gartner S, y Grupo de Trabajo de Fibrosis Quística. Protocolo de diagnóstico y seguimiento de los pacientes con fibrosis quística. An Pediatr (Barc). 2009;71:250-64.
- Largo García I. Fibrosis Quística. Rev Ped Elec. 2009;6(1) [en línea]. Disponible en: http://www.revistapediatria.cl/vol6num1/pdf/2_FIBROSIS_QUISTICA.pdf