


Tratamiento con hormona de crecimiento: indicaciones y aspectos prácticos para la consulta de Atención Primaria
2 Servicio de Endocrinología Pediátrica. Hospital Virgen de la Salud. Toledo. (España).
3 Servicio de Endocrinología Pediátrica. Hospital Virgen de la Salud. Toledo. (España).
RESUMEN
Los trastornos del crecimiento en la infancia son uno de los motivos de consulta más frecuentes en Pediatría. El pediatra de Atención Primaria vigila a diario el crecimiento de los niños, por lo que su papel es crucial en la detección precoz de la talla baja.
Conocer las indicaciones y criterios del tratamiento con hormona de crecimiento recombinante (rGH) puede ayudar a identificar mejor a los posibles candidatos, adelantando el inicio del tratamiento y mejorando así la respuesta.
Además, en los últimos años han sido aceptadas nuevas indicaciones de tratamiento, por lo que actualmente podrían existir pacientes con talla baja, que no cumplían criterios de tratamiento previamente, que podrían beneficiarse del mismo.
INTRODUCCIÓN
La hormona de crecimiento (GH) o somatotropina es una proteína secretada a nivel hipofisario por las células somatotropas; ejerce su acción principal sobre el hueso y tejido conectivo, estimulando el crecimiento longitudinal durante la infancia. Asimismo, posee acciones metabólicas como la estimulación de la lipólisis y la síntesis proteica, ejerce una acción antiinsulínica y mejora la mineralización ósea, entre otras.
La secreción de GH por la adenohipófisis es pulsátil: se alcanzan los picos más altos durante el sueño profundo y puede llegar a ser completamente indetectable en otros momentos del día. La secreción de GH se encuentra regulada por un complejo sistema, pudiendo estimularse en casos de estrés, hipoglucemia, hipotensión o ejercicio.
Los niños que presenten un estancamiento de la talla o de la velocidad de crecimiento, talla inferior a la talla genética o talla en -2 desviaciones estándar (DE) deben ser estudiados. El estudio de la talla debe realizarse con curvas de crecimiento actualizadas, en función del sexo y la etnia, utilizando las curvas de estudios transversales para mediciones aisladas, y de estudios longitudinales para poder valorar la velocidad del crecimiento.
La valoración inicial del niño con talla baja debe incluir una anamnesis exhaustiva, una exploración física completa y estudios analíticos y radiológicos para poder descartar otras causas de talla baja como anemia, hipotiroidismo, diabetes, displasias óseas, raquitismo, enfermedades renales o patologías metabólicas (Tabla 1).
Tabla 1. Estudios complementarios en el niño con talla baja. Mostrar/ocultar

El objetivo de este artículo es revisar las indicaciones actuales de tratamiento con GH, así como los puntos claves del seguimiento de estos pacientes que día a día pasan por nuestras consultas.
INDICACIONES DE TRATAMIENTO CON HORMONA DE CRECIMIENTO
La hormona de crecimiento se utiliza como tratamiento desde mediados del siglo XX. Inicialmente se extraía de hipófisis humanas, y la escasa disponibilidad condicionaba que se limitara su uso únicamente al déficit severo de GH. Los avances en las técnicas genéticas permitieron el desarrollo de la hormona de crecimiento recombinante (rGH), idéntica a la natural, lo que optimizó los tratamientos y permitió el estudio y aplicación en otras patologías.
Las indicaciones aprobadas actualmente por la Agencia Española del Medicamento y Productos Sanitarios (AEMPS) para el tratamiento con hormona de crecimiento recombinante1 son:
- Déficit clásico o defecto de hormona de crecimiento.
- Trastorno del crecimiento asociado a síndrome de Turner.
- Trastorno del crecimiento asociado a niños nacidos pequeños para su edad gestacional (PEG), sin crecimiento recuperador.
- Síndrome de Prader-Willi.
- Trastorno del crecimiento asociado a insuficiencia renal crónica.
- Deficiencia de crecimiento asociado a alteración en el gen SHOX.
En otros países, además de estas indicaciones, el tratamiento ha sido aprobado para niños con talla baja idiopática y niños con trastorno de crecimiento asociado a síndrome de Noonan, pero de momento estas indicaciones no han sido aprobadas en España.
La rGH se dispensa en las indicaciones autorizadas por el ministerio, con cargo a los presupuestos de la Sanidad Pública, pero se exige para ello el diagnóstico hospitalario y la aprobación por parte de un comité de expertos (nacional o autonómico) de un protocolo de tratamiento, con consentimiento informado y revisión anual.
Deficit clásico de hormona de crecimiento
El déficit clásico de hormona de crecimiento fue una de las primeras indicaciones de tratamiento con GH. Numerosos estudios han observado mejoría de la talla final en estos niños, especialmente cuando el tratamiento se inicia de manera precoz2.
El diagnóstico del déficit de hormona de crecimiento se basa en parámetros auxológicos y estudios analíticos. En ocasiones, el déficit clásico puede debutar en periodo neonatal con clínica de hipoglucemia, ictericia prolongada y malformaciones genitales, aunque en la mayoría de los casos se manifiesta como deterioro progresivo de la talla en los primeros años de vida.
El déficit de GH neonatal no precisa del cumplimiento de ningún requisito auxológico para iniciar el tratamiento; el inicio precoz del mismo no solo mejora la talla final del paciente, sino que ayuda al manejo inicial de la clínica metabólica asociada como la hipoglucemia3.
La característica clínica del déficit de GH de aparición más tardía es el retraso del crecimiento. Los criterios definidos por el comité asesor para poder iniciar el tratamiento en este caso sí que son fundamentalmente auxológicos4.
- Talla inferior a -2 DE o inferior a 1 DE de la talla media parental y, en su caso, predicción de talla adulta inferior a la talla genética en más de 1 DE.
- Velocidad de crecimiento disminuida de manera mantenida, durante al menos seis meses, encontrándose por debajo del percentil 10 para su edad ósea correspondiente.
- Retraso de la maduración ósea en más de un año con respecto a la edad cronológica, salvo en el caso de que se asocie a pubertad precoz central secundaria a radioterapia.
En caso de sospecha clínica de déficit de GH se debe determinar la somatomedina C (IGF-1) y la proteína transportadora (IGFBP-3). La IGF-1 es un polipéptido que producen algunos tejidos, como el hígado, en respuesta a la GH hipofisaria. Una vez sintetizada, forma un complejo en el plasma al unirse a la IGFBP-3 y la subunidad ácido lábil (ALS) para poder ejercer su acción en los diferentes tejidos. A diferencia de la hormona de crecimiento, la IGF-1 y la IGFBP-3 no tienen una secreción pulsátil, lo que facilita su interpretación. Cuando sus cifras se encuentran disminuidas, se debe sospechar un déficit de GH siempre que se descarten otros factores que en ocasiones pueden modificarlas como la malnutrición, el hipotiroidismo o algunas hepatopatías.
Finalmente, para completar el estudio, se deben realizar pruebas de estimulación de GH que nos permitan valorar el pico máximo de secreción de la hormona, resultando patológicos en caso de presentar picos de GH inferiores a los valores de referencia según el laboratorio. Una vez confirmado el déficit de GH, siempre se debe realizar una prueba de imagen que informe del estado de la hipófisis, ya que en muchos casos esta se puede encontrar alterada (ectopia, hipoplasia, agenesia, lesión tumoral…), justificando así la secreción patológica de la hormona.
Trastorno de crecimiento asociado a síndrome de Turner
El síndrome de Turner es un trastorno cromosómico producido por la monosomía total o parcial del cromosoma X.
La presentación clínica es muy variable: en ocasiones se detecta en periodo prenatal, y en otras puede pasar desapercibida hasta la adolescencia, donde se manifiesta como amenorrea primaria.
Las manifestaciones más características de este síndrome, además del fenotipo típico (pterigium colli, tórax ancho, mamilas separadas, inserción baja de pelo, cubitus valgo, hábito pseudohercúleo, alteraciones dentales y ungueales…), en ocasiones poco llamativo, son la disgenesia gonadal y la talla baja. Esta última se cree que puede ser debida a la haploinsuficiencia del gen SHOX, entre otras causas.
La frecuencia de este síndrome hace que sea recomendable realizar un cariotipo en toda niña con talla baja sin otra causa diagnosticada, para descartarlo aunque el fenotipo no lo sugiera.
El retraso de crecimiento asociado al síndrome de Turner se puede beneficiar del tratamiento con rGH. Se estima que estas niñas podrían presentar una ganancia de unos 7-10 cm al recibir el tratamiento en comparación con las que no lo reciben5. Algunos estudios, además, han observado que el inicio precoz a partir de los dos años de edad, consigue mejores resultados de talla final que el tratamiento a altas dosis posteriormente. Por ello, se debe valorar el inicio del tratamiento tras el diagnóstico citogenético, a partir de los dos años de vida, si presenta retraso de crecimiento durante al menos seis meses.
Retraso del crecimiento asociado a insuficiencia renal crónica
El retraso de crecimiento asociado a la insuficiencia renal crónica (IRC) se relaciona con múltiples factores etiológicos, entre los que se encuentran la propia enfermedad renal, la inadecuada nutrición, la osteodistrofia renal, la acidosis metabólica y la insensibilidad a la GH.
Algunos estudios han observado que el tratamiento con rGH mejora considerablemente la talla en estos pacientes, observándose una mejoría de la talla y velocidad de crecimiento, especialmente en los primeros dos años de tratamiento, pudiendo llegar a alcanzar tallas adultas dentro de la normalidad6.
Por esta razón estaría indicado el inicio de tratamiento en niños con insuficiencia renal crónica, a partir de los dos años de edad, y en estado prepuberal, que se encuentren en tratamiento crónico de diálisis, siempre que presenten un retraso de crecimiento diagnosticado.
Previo al inicio del tratamiento se debe optimizar el estado nutricional, hidroelectrolítico, y metabólico, y haber intentado disminuir la dosis de corticoides a la mínima eficaz.
En los niños con IRC que finalmente reciben un trasplante renal, el tratamiento con rGH debe ser suspendido, y en caso de que persista el retraso del crecimiento, a pesar de tener una función renal normal, se puede valorar el reinicio del tratamiento, ya que se ha observado una mejoría de la talla posterior6.
En algunos casos, el tratamiento de estos niños se ha relacionado con un aumento de la incidencia de hipertensión intracraneal idiopática, epifisiolisis de la cabeza femoral y empeoramiento de escoliosis ya existente. Sin embargo, no se ha observado empeoramiento o repercusión sobre la función renal, aunque en ocasiones pueden aumentar ligeramente los niveles de creatinina durante el tratamiento7.
El inicio del tratamiento estaría especialmente contraindicado en estos niños en caso de patología cardiovascular severa, osteopatía severa, diabetes mellitus manifiesta, enfermedad maligna activa, o trasplante renal.
Síndrome de Prader-Willi
El síndrome de Prader-Willi es la forma de obesidad sindrómica más frecuente en la infancia. Está producido por una inactivación o pérdida de los genes de la región15q11-q13 heredados del padre, que no pueden ser complementados por genes maternos, ya que se encuentran silenciados por un mecanismo de imprinting genético.
Los niños con síndrome de Prader-Willi pueden presentar clínica ya en el periodo prenatal, con disminución de los movimientos fetales, que en muchas ocasiones produce presentaciones podálicas. Posteriormente, en el periodo neonatal, se comienza a observar hipotonía severa, criptorquidia o clítoris hipoplásico, y problemas para la alimentación, dando lugar a un fallo de medro progresivo en los primeros meses de vida. En la infancia precoz, comienza la hiperfagia compulsiva, produciendo una ganancia ponderal importante y obesidad, lo que se acompaña en algunos casos del deterioro progresivo de la talla.
La variabilidad clínica en función de la edad llevó a Holm en 1993 a unificar unos criterios diagnósticos clínicos (Tabla 2) para orientar el diagnóstico y valorar en qué casos se debería solicitar estudio genético para confirmar la enfermedad8.
Tabla 2. Criterios clínicos diagnósticos del síndrome de Prader-Willi Mostrar/ocultar

El síndrome de Prader–Willi es una de las indicaciones aceptadas para el tratamiento con rGH. Los objetivos del tratamiento difieren en parte de los de las otras indicaciones, ya que en este caso los beneficios de la hormona de crecimiento no residen tan solo en el crecimiento longitudinal, sino también en ayudar a mejorar la composición corporal en cuanto a la grasa y la densidad mineral ósea9. Por este motivo, el tratamiento con rGH, estaría indicado en estos niños tras la confirmación genética, a partir de los dos años de vida.
El tratamiento con rGH en el síndrome de Prader-Willi se debe realizar con especial precaución, debido a que se han descrito un mayor número de complicaciones, especialmente respiratorias, que suelen aparecer en las primeras semanas de tratamiento. El inicio o la continuidad del tratamiento estarían contraindicados en caso de obesidad mórbida, diabetes mellitus o intolerancia a la glucosa, síndrome de apnea del sueño, hipertrofia obstructiva amigdaloadenoidea, escoliosis mayor de 20°, enfermedad aguda, proceso tumoral activo o edad ósea adulta.
La mayor incidencia de complicaciones, justifica que antes de iniciar el tratamiento sea preciso, además de realizar una analítica completa y estudios básicos como en el resto de las indicaciones, realizar un estudio polisomnográfico, estudio de composición corporal mediante densitometría ósea (DXA) o impedanciometría, estudio radiológico de columna, y test de tolerancia a la glucosa.
Una vez iniciado el tratamiento, se realizarán controles periódicos como en las otras indicaciones, pero además se debe tener un estricto control de la posible aparición de síndrome de apnea-hipopnea obstructiva del sueño (SAHOS), alteración del perfil lipídico o glucémico, así como empeoramiento de la obesidad.
Trastorno del crecimiento en niños nacidos pequeños para la edad gestacional (PEG)
Un niño es considerado PEG cuando presenta una longitud y/o peso al nacimiento igual o inferior a -2 DE para su población de referencia, edad gestacional y sexo.
La mayoría de estos niños presentan un crecimiento recuperador, también denominado catch-up, en los primeros dos años de vida, alcanzando el percentil 3 a esa edad, y pudiendo llegar a alcanzar una talla adulta normal, aunque generalmente 1 DE inferior a lo que les correspondería10. Sin embargo, hasta un 10% de estos niños no presentan ese crecimiento recuperador y, en el caso de los PEG que además son prematuros, puede verse retrasado.
El tratamiento con hormona de crecimiento debe ser considerado cuando un niño nacido PEG que no presenta un crecimiento recuperador en los primeros cuatro años de vida, se mantiene con una talla inferior a -2,5 DE y/o menor de -1 DE ajustada a la talla diana.
El tratamiento con rGH produce una mejoría en la talla final, siendo necesarias dosis mayores que las empleadas en el déficit clásico de GH, ya que se piensa que podría existir una cierta resistencia relativa a la hormona y sus derivados.
Previo al inicio del tratamiento es necesario descartar cuadros sindrómicos asociados a los niños PEG, como pueden ser el síndrome de Silver-Russell, así como trastornos del metabolismo hidrocarbonado (diabetes mellitus, resistencia insulínica o intolerancia hidrocarbonada) ya que existe una mayor predisposición en estos niños, aunque se ha visto que ese efecto es reversible al suspender el tratamiento. Por este motivo, es fundamental, además del adecuado control auxológico, realizar controles periódicos de presión arterial, y controles analíticos anuales que incluyan glucemia e insulinemia basales, glicohemoglobina, lipidograma, y perfil tiroideo.
Retraso del crecimiento asociado a alteración del gen SHOX
El genshort stature homeobox (SHOX) se localiza en la región pseudoautosómica 1 oPAR1 del brazo corto de ambos cromosomas sexuales (X e Y), escapándose a la inactivación del cromosoma X, y sufriendo recombinación durante la meiosis11.
La clínica derivada de la alteración del gen SHOX afecta fundamentalmente a la talla y es dosis dependiente; por ello, en función de la cantidad del material genético alterado, el espectro y la gravedad pueden ser variables. Algunos de estos pacientes pueden presentar exclusivamente talla baja, otros alteraciones radiológicas características de la discondrosteosis de Léri-Weill, existiendo además formas graves como la displasia mesomélica de Langer, en la que existe alteración de los dos alelos del gen SHOX11.
La alteración del gen SHOX se encuentra en alrededor del 2-15 % de los pacientes diagnosticados previamente de talla baja idiopática. Muchos se diagnostican de manera tardía, especialmente cuando no existen signos radiológicos que lo sugieran, ya que las deformidades son evolutivas y pueden no estar presentes en los primeros años de vida. Por ello, debemos sospechar alteración del gen SHOX en pacientes con talla baja familiar con patrón de herencia autosómica dominante.
El retraso del crecimiento asociado al gen SHOX fue una de las últimas indicaciones aprobadas para el tratamiento con rGH. Tras el descubrimiento del gen, se pensó que estos pacientes debían mejorar al recibir tratamiento con rGH, al igual que ocurría en las niñas con síndrome de Turner.
Varios estudios posteriores mostraron que el crecimiento en niños con alteración del gen SHOX mejoraba con este tratamiento, incrementándose la velocidad de crecimiento y aumentando la talla final, observándose resultados similares en estos pacientes y en las niñas con síndrome de Turner12.
El tratamiento con rGH estaría indicado tras el estudio de genética molecular y la confirmación de la mutación del gen, siempre que el paciente sea mayor de dos años de edad y presente un retraso de crecimiento.
Otras indicaciones
El tratamiento de rGH se utiliza en otros países con otras indicaciones, entre las que se encuentran los niños con síndrome de Noonan y los pacientes con talla baja idiopática (TBI), una de las indicaciones más polémicas. En España, de momento, no está autorizado su uso para estas indicaciones.
La TBI engloba a un gran grupo de los pacientes con talla baja, en los que no se encuentra ninguna causa que lo justifique. En 2003 la Food and Drug Administration (FDA) aprobó el uso de rGH en estos niños cuando presentan una talla en -2,25 DE y una velocidad de crecimiento enlentecida. Los estudios existentes sobre los beneficios de la rGH en la TBI aún son escasos; algunos sí que observan ganancias en la talla a corto plazo y en la talla final, aunque la talla adulta alcanzada generalmente se sigue manteniendo en el límite bajo de la normalidad13,14.
El tratamiento del síndrome de Noonan ha sido aprobado en EE. UU. por laFDA, con estudios que sugieren una mejoría de la velocidad de crecimiento y una mayor ganancia de talla en comparación con los niños no tratados, aunque los estudios son aún escasos y a corto plazo14,15.
DOSIS DE rGH SEGÚN LA INDICACIÓN
La hormona de crecimiento recombinante se administra en todos los casos de manera subcutánea y en una única dosis nocturna.
La dosis varía de una indicación a otra, dosificándose habitualmente en función del peso, excepto si existe obesidad o síndrome de Turner, en las que se suele hacer en función de la superficie corporal (Tabla 3).
Tabla 3. Dosificación del tratamiento con GHr Mostrar/ocultar
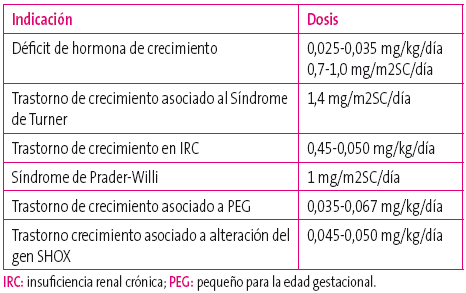
La dosis de rGH se irá modificando periódicamente basándose en la velocidad de crecimiento, los niveles de IGF-1 e IGFBP-3, o la aparición de algún efecto secundario o contraindicación, que pueda condicionar la disminución de la dosis o incluso la suspensión del tratamiento.
En caso de sospecha de no respuesta al tratamiento, se debe tener en cuenta la dosis administrada, revisar la técnica de administración y especialmente la adhesión al tratamiento, una de las causas más frecuentes de no respuesta. En caso de que todo esté correcto, se debe valorar el aumento de la dosis y en caso de no mejoría, el tratamiento debería ser suspendido.
SEGUIMIENTO Y CONTROLES EN LOS PACIENTES TRATADOS
Todos los pacientes en tratamiento con rGH deben realizar controles periódicos de peso, talla y velocidad de crecimiento al menos cada 3-6 meses, así como toma de presión arterial.
Anualmente se debe realizar estudio analítico para controlar especialmente los niveles de IGF-1 e IGFBP-3, el perfil tiroideo para descartar la aparición de hipotiroidismo y el perfil glucémico.
Además se debe vigilar que no exista una aceleración excesiva de la edad ósea que pueda perjudicar el efecto sobre la talla final, por lo que se solicitará anualmente un estudio radiológico.
En caso de dolor de espalda o signos de escoliosis se debe solicitar estudio radiológico de la estática de la columna dorsolumbar, ya que el tratamiento puede agravar una escoliosis previa.
Los pacientes con síndrome de Prader-Willi, además de los controles habituales, deben tener un seguimiento de la composición corporal y la posible aparición de un SAHOS.
El tratamiento se podrá mantener hasta que la velocidad de crecimiento disminuya (< 2 cm/año) y/o se alcance la edad ósea adulta, aunque deberá ser retirado siempre que aparezca alguna contraindicación.
El déficit clásico de GH es la única indicación en la que los pacientes, al alcanzar la edad adulta, deben ser reevaluados para valorar la continuidad del tratamiento como tratamiento sustitutivo.
EFECTOS SECUNDARIOS DEL TRATAMIENTO CON rGH
Los riesgos derivados del tratamiento con GH son escasos e infrecuentes. Sin embargo, la experiencia de seguridad del tratamiento se basa en niños con déficit clásico de GH, siendo escasos los estudios en niños sin déficit, en los que los niveles de GH pueden llegar a duplicar o triplicar la producción endógena normal. Por ello, se debe ser cauto a la hora de valorar los riesgos del tratamiento a medio y largo plazo.
Los efectos secundarios descritos actualmente en los pacientes en tratamiento con rGH son la hipertensión intracraneal idiopática, cefalea, convulsiones, dolores musculares y artralgias, empeoramiento de la escoliosis, epifisiolisis, alteración del metabolismo hidrocarbonado por su efecto antiinsulínico, hipertrigliceridemia, hipotiroidismo, recidiva tumoral de tumor previo, reacciones locales en zona de inyección, exacerbaciones de psoriasis y dermatitis, y desarrollo de anticuerpos frente a la somatotropina1,16.
CONTRAINDICACIONES DEL TRATAMIENTO
La rGH está contraindicada siempre que exista una hipersensibilidad a la hormona, edad ósea adulta con epífisis cerradas, enfermedad tumoral activa o recidiva o progresión de una lesión intracraneal previa, enfermedad aguda crítica, especialmente tras cirugías cardiacas o abdominales, fallo respiratorio, retinopatía diabética proliferativa o no proliferativa severa, y en los pacientes con síndrome de Prader-Willi que presenten obesidad severa, o síndrome de apnea obstructiva del sueño1,16.
CUADERNO DEL PEDIATRA
- Se debe revisar periódicamente el crecimiento de todos los niños y realizarse un estudio en caso de estancamiento de la talla o la velocidad de crecimiento, talla en -2 DE o talla inferior a la talla genética.
- El pediatra debe conocer las indicaciones de tratamiento con hormona de crecimiento para derivar lo antes posible a los pacientes y mejorar así la respuesta al mismo.
- En toda niña con retraso de crecimiento y estudio inicial normal se recomienda solicitar un cariotipo para descartar un síndrome de Turner.
- La talla baja asociada a obesidad, especialmente si se asocia a edad ósea retrasada, debe hacer sospechar una patología subyacente, como el síndrome de Prader-Willi.
- Los niños nacidos pequeños para la edad gestacional con talla baja pueden beneficiarse de tratamiento en caso de no presentar crecimiento recuperador a los cuatro años.
- La alteración del gen SHOX se debe sospechar en pacientes con talla baja familiar de herencia autosómica dominante.
- La rGH se administra subcutáneamente en una dosis única diaria, nocturna.
- En caso de no respuesta al tratamiento se debe revisar la técnica de administración, vigilar la adherencia al tratamiento y valorar el aumento de la dosis.
- El tratamiento será suspendido cuando no exista respuesta, la velocidad de crecimiento disminuya, se alcance edad ósea adulta o aparezca alguna contraindicación.
BIBLIOGRAFÍA
- Somatropina. En: Pediamécum [en línea] [consultado el 03/09/2015]. Disponible en: http://pediamecum.es/somatropina/
- Rogol AD. Treatment of growth hormone deficiency in children. En: UpToDate [en línea] [consultado el 03/09/2015]. Disponible en: http://www.uptodate.com/contents/treatment-of-growth-hormone-deficiency-in-children
- Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (gh) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocrinol Metab. 2000;85:3990-3.
- Comité Asesor para la Hormona de Crecimiento del Ministerio de Sanidad y Consumo. Criterios para la utilización racional de la hormona de crecimiento en niños. En: Ministerio de Sanidad y Consumo [en línea] [consultado el 03/09/2015]. Disponible en: http://www.msssi.gob.es/profesionales/farmacia/pdf/criteriosHCninos020908.pdf
- Baxter L, Bryant J, Cave CB, Milne R. Recombinant growth hormone for children and adolescents with Turner syndrome. Cochrane Database Syst Rev. 2007(1):CD003887.
- Hodson EM, Willis NS, Craig JC. Growth hormone for children with chronic kidney disease. Cochrane Database Syst Rev. 2012;2:CD003264.
- Tönshoff B. Growth hormone treatment in children with chronic kidney disease and postrenal transplantation. En: UpToDate [en línea] [consultado el 03/09/2015]. Disponible en: http://www.uptodate.com/contents/growth-hormone-treatment-in-children-with-chronic-kidney-disease-and-postrenal-transplantation
- Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91:398-402.
- Bakker NE, Kuppens RJ, Siemensma EP, Tummers-de Lind van Wijngaarden RF, Festen DA, Bindels-de Heus GC, et al. Eight years of growth hormone treatment in children with Prader-Willi syndrome: maintaining the positive effects. J Clin Endocrinol Metab. 2013;98:4013-22.
- Díez Lópeza I, de Arriba Muñoz A, Bosch Muñoz J, Cabanas Rodríguez P, Gallego Gómez E, Martínez-Aedo Ollero MJ, et al. Pautas para el seguimiento clínico del niño pequeño para la edad gestacional. An Pediatr. 2012;76:104.e1-7
- Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011;75:81-9.
- Blum WF, Ross JL, Zimmermann AG, Quigley CA, Child CJ, Kalifa G, et al. GH treatment to final height produces similar height gains in patients with SHOX deficiency and Turner syndrome: results of a multicenter trial. J Clin Endocrinol Metab. 2013;98:E1383-92.
- Bryant J, Baxter L, Cave CB, Milne R. Recombinant growth hormone for idiopathic short stature in children and adolescents. Cochrane Database Syst Rev. 2007(3):CD004440.
- Loche S, Carta L, Ibba A, Guzzetti C. Growth hormone treatment in non-growth hormone-deficient children. Ann Pediatr Endocrinol Metab. 2014;19:1-7.
- Giacomozzi C, Deodati A, Shaikh MG, Ahmed SF, Cianfarani S. The impact of growth hormone therapy on adult height in Noonan syndrome: A systematic review. Horm Res Paediatr. 2015;83:167-76.
- Waltham MA, Aragonés Gallego A. Recombinant human growth hormone (somatropin): Pediatric drug information. En: UpToDate [en línea] [consultado el 03/09/2015]. Disponible en: http://www.uptodate.com/contents/recombinant-human-growth-hormone-somatropin-pediatric-drug-information
LECTURAs RECOMENDADAs
-
Loche S, Carta L, Ibba A, Guzzetti C. Growth hormone treatment in non-growth hormone-deficient children. Ann Pediatr Endocrinol Metab. 2014 Mar;19:1-7.
Revisión actualizada de cada una de las indicaciones en las que no existe déficit de GH.
-
López Siguero, J.P. Talla baja idiopática y hormona de crecimiento: bastantes dudas y algunas recomendaciones. Evid Pediatr. 2011,7:51.
Revisión de la talla baja idiopática y su tratamiento con hormona crecimiento en otros países.