



Dolor abdominal en hijo de padres nigerianos: ¿podría tratarse de drepanocitosis?
2 Hospital Materno Infantil. Málaga. (España).
PUNTOS CLAVE
- El dolor abdominal es un síntoma inespecífico que plantea un diagnóstico diferencial amplio.
- Una posible causa de dolor abdominal en niños en edad escolar es la drepanocitosis.
- Clasificar la anemia en función de la respuesta reticulocitaria y el tamaño del hematíe podría ser una buena estrategia diagnóstica.
- En la mayoría de los casos, la hemólisis suele tener un componente mixto, tanto intravascular como extravascular.
- En las anemias regenerativas, independientemente de la causa, debido a la eritropoyesis aumentada, es fundalmente el aporte de folatos.
- La hemoglobinopatía SC es una forma leve de drepanocitosis que habitualmente pasa desapercibida en la infancia temprana.
- El manejo de la drepanocitosis es más fácil si se conocen los motivos de consulta más habituales en las descompensaciones de esta, aunque en muchos casos el diferenciarlos de cuadros intercurrentes plantea un reto diagnóstico y terapéutico.
- En los pacientes estables se realizará un seguimiento semestral por Hematología, mientras que en los inestables se llevará a cabo un seguimiento más frecuente.
RESUMEN
En un paciente de raza negra con dolor abdominal en edad escolar hay que plantear la drepanocitosis como posible diagnóstico. La ecografía abdominal y el hemograma centrarán la sospecha, que será confirmada mediante la electroforesis de hemoglobina. El tratamiento de mantenimiento se basa en la administración de folatos y ante la aparición de fiebre, dolores persistentes torácicos, abdominales u óseos, síntomas neurológicos o priapismo, debe remitirse a un Servicio de Urgencias Hospitalarias para descartar complicaciones. El seguimiento por Hematología y Oftalmología se realizará de forma estrecha en los casos inestables y de forma semestral si no presentan complicaciones. En la infancia temprana, una buena estrategia sería hacer coincidir sus revisiones con las vacunaciones1. Los pacientes afectos de esta enfermedad suelen tener una buena evolución clínica con el seguimiento y recomendaciones oportunos.
CASO CLÍNICO
Un niño varón de raza negra, de nueve años de edad, acude a la consulta de Urgencias de su centro de salud por presentar un cuadro de ocho horas de dolor abdominal localizado en hemiabdomen izquierdo. No asociaba diarrea, alteraciones miccionales, sintomatología respiratoria ni otra clínica acompañante. Tampoco refería encontrarse en un ambiente epidemiológico infeccioso ni mencionaba toma de fármacos o alimentos no habituales en los días previos.
En la exploración mantenía un estado general conservado, presentaba palidez mucosa y frecuencia cardiaca de 100 latidos por minuto, encontrándose afebril, con auscultación cardiopulmonar normal. La palpación abdominal se encontraba dificultada por el dolor, con sensación de puñopercusión renal positiva. No existían otros datos de interés.
Como antecedentes relevantes, había padecido una faringoamigdalitis aguda vírica hacía dos semanas. Aunque sus padres eran de nacionalidad nigeriana, había nacido en España y nunca había viajado al extranjero. Estaba vacunado según el calendario de Andalucía, y no se le conocían alergias. Tenía tres hermanas, una de ellas transfundida en su país de origen por un cuadro febril a los dos años de edad (desafortunadamente sin más información al respecto).
En la consulta se realizó tira reactiva en orina, detectándose únicamente presencia de cuerpos cetónicos, y, ante la persistencia del dolor tras la administración de analgesia, se deriva al hospital terciario de referencia para una evaluación más completa.
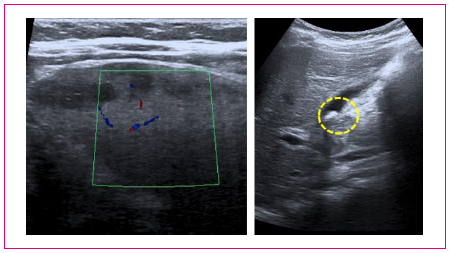
Cuatro horas más tarde, al ser revalorado en el Servicio de Urgencias Hospitalarias, refiere aparición de vómitos de contenido gástrico y fiebre de hasta 38,5 °C, persistiendo una exploración similar a la previa. Se realizó una radiografía de tórax, analítica de sangre y ecografía abdominal. En el hemograma existía anemia microcítica-hipocroma, reticulocitosis y leve plaquetopenia; la serie blanca era normal. La bioquímica y gasometría fueron normales, a excepción de una leve elevación de proteína C reactiva y la lactato-deshidrogenasa, siendo normales los perfiles de función renal, hepática y vía biliar (bilirrubina total y directa normales). La ecografía Doppler abdominal mostró un bazo aumentado de tamaño, con áreas hipoecoicas en la periferia que podrían corresponder a zonas hipoperfundidas, además de una colelitiasis sin signos de colecistitis (Figura 1). La radiografía de tórax fue normal.
Figura 1. Ecografía abdominal. A la izquierda se muestra la ecoestructura heterogénea del bazo, a la derecha el contenido litiásico de la vesícula biliar Mostrar/ocultar
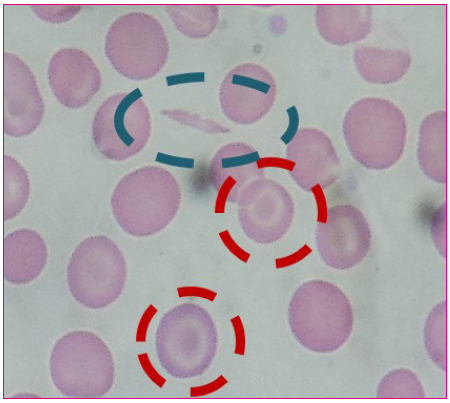
Debido a estos hallazgos, ingresa en planta para estudio, donde se amplían las pruebas complementarias con perfil férrico y velocidad de sedimentación globular, que fueron normales, además de un test de Coombs directo con resultado negativo. En el frotis de sangre periférica se encontraron dianocitos, hematíes plegados y algún drepanocito (Figura 2), lo que, junto con la realización de una electroforesis de hemoglobinas, facilitó el diagnóstico. Esta prueba reveló la presencia de bandas correspondientes a cadenas S y C, que nos arroja el diagnóstico de anemia falciforme SC, compatible con la sospecha clínica.
Figura 2. Frotis de sangre periférica. En el círculo azul se engloba un drepanocito y en los rojos dianocitos Mostrar/ocultar
DISCUSIÓN
Diagnóstico diferencial del dolor abdominal
El dolor abdominal es el síntoma por el que consulta el paciente, y a partir de él se planteó el diagnóstico diferencial. Este puede aparecer por alteración de estructuras intraabdominales –bien de abordaje médico o que requieran de actuación quirúrgica urgente– o causas extraabdominales.
Las causas intraabdominales con abordaje quirúrgico se podrían agrupar en cinco grandes síndromes –peritoneal, oclusivo, hemorrágico, torsional y perforativo–, siendo las causas más frecuentes la apendicitis aguda, la invaginación intestinal, la úlcera péptica, la rotura visceral o la estrangulación de estructuras. Entre los cuadros de abordaje médico más probables encontraríamos la gastroenteritis, la pielonefritis, la colecistitis y la hepatitis agudas2.
Otras patologías propias de estructuras no abdominales también pueden causar dolor abdominal, como son la neumonía –en muchos casos desapercibida por su presentación con este síntoma–, alteraciones hematológicas, endocrino-metabólicas, como la cetoacidosis diabética, reumatológicas y vasculares e incluso con foco infeccioso extraabdominal, como la faringoamigdalitis aguda2.
Diagnóstico diferencial de la anemia
Las causas de la anemia (Figura 3 y Tabla 1) se pueden dividir entre aquellas con respuesta reticulocitaria, periféricas o regenerativas y las que no la tienen, centrales o arregenerativas. En este caso nos centraremos en las regenerativas, causadas por hemorragias o bien por hemólisis, basadas o no en un mecanismo inmunomediado.
Figura 3. Clasificación general de las anemias Mostrar/ocultar
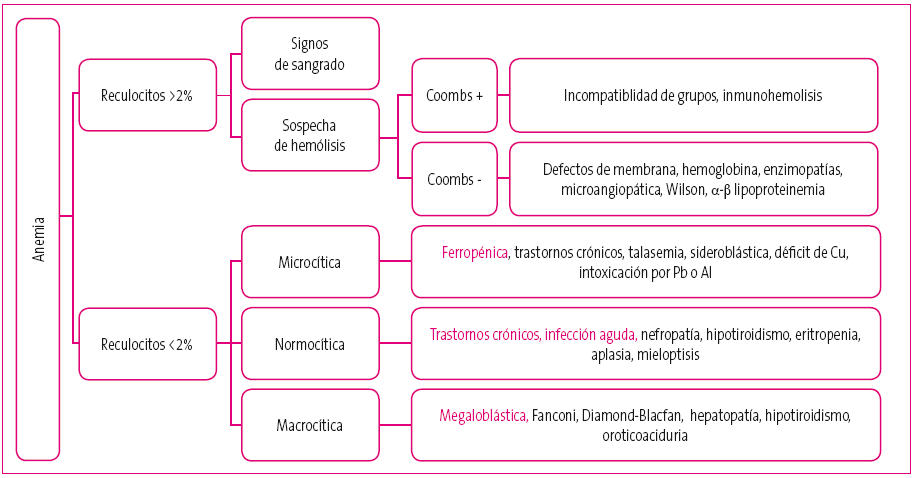
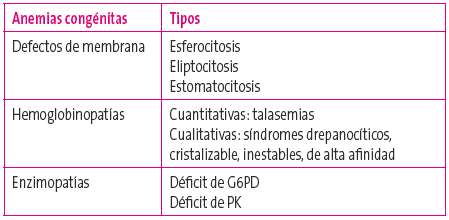
Tabla 1. Clasificación de los defectos corpusculares del hematíe Mostrar/ocultar
Entre las anemias arregenerativas microcíticas encontramos la ferropenia, causa más frecuente de anemia en la infancia3,4.
La hemólisis puede ser de predominio intravascular, cuando los hematíes se rompen en el torrente circulatorio, o extravascular, si se rompen en el bazo o el hígado. La rotura intravascular del hematíe se produce por alteraciones del mismo o bien por elementos externos que causan su rotura, provocando la liberación de la hemoglobina al torrente circulatorio, que, por ser un tóxico se une a la haptoglobina y la hemopexina, entre otras globulinas, descendiendo sus niveles. Si la hemolisis es muy intensa, la hemoglobina se acaba filtrando por el riñón produciendo hemoglobinuria y hematuria, y más tardíamente hemosiderinuria.
La rotura extravascular se produce en las localizaciones de hemocatéresis fisiológica, puesto que en este caso el problema es la escasa resistencia de la membrana del eritrocito al estrés mecánico, con supervivencia celular acortada. En estas vísceras es el sistema fagocítico mononuclear el encargado de degradar la hemoglobina, que pasará al sistema porta como bilirrubina no conjugada y, si excede la capacidad enzimática del hígado, elevará la bilirrubina indirecta en sangre periférica, pudiendo causar ictericia acolúrica. Una hemólisis crónica podría producir cálculos biliares de bilirrubinato cálcico.
Esta división es puramente académica, puesto que toda hemólisis consta de ambos componentes, siendo los únicos datos diferenciales y exclusivos la presencia de hemoglobina libre, hemoglobinuria y hemosiderinuria.
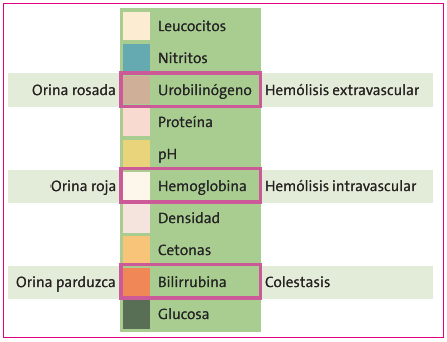
Es interesante conocer que la coloración de la orina puede orientarnos hacia cada proceso (Figura 4). Las orinas rosadas se deben a la presencia de urobilinógeno producido en la hemólisis extravascular; las orinas rojas, a la hemoglobina y por lo tanto intravascular o nefrourológica, y la orina parduzca o coluria a la bilirrubina directa o conjugada, que solo puede detectarse en orina en los cuadros colestásicos. Así podemos entender que la ictericia de causa hematológica no puede producir coluria puesto que la bilirrubina indirecta no se filtra por el riñón3,4.
Figura 4. Hallazgos comparativos entre el aspecto de la orina, tira reactiva y proceso desencadenante Mostrar/ocultar
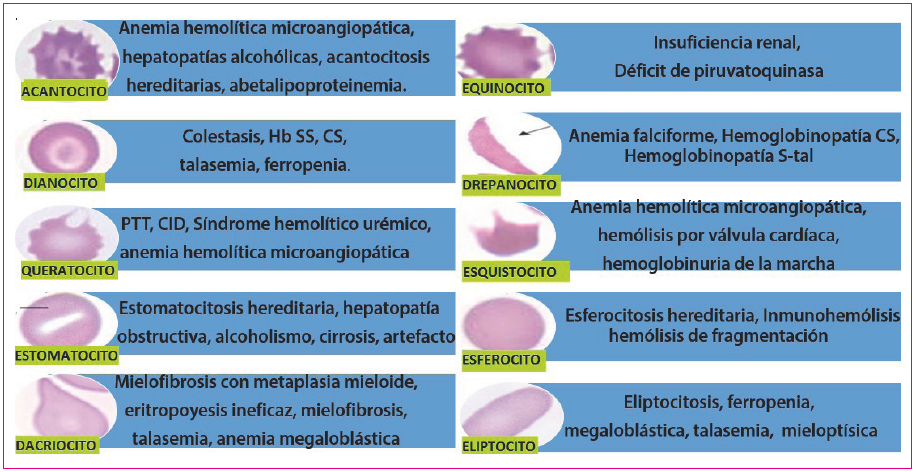
Mediante el frotis de sangre periférica pueden detectarse algunas de las causas de hemólisis intravascular no inmune puesto que se asocian a formas anómalas del hematíe que, si bien no son específicas, son muy sugestivas de algunas de estas patologías (Figura 5)4.
Figura 5. Morfologías del hematíe y patologías en las que es más frecuente su aparición Mostrar/ocultar
HEMOGLOBINOPATÍA SC
Epidemiología
La enfermedad de la hemoglobina SC, o drepanocitosis en heterocigosis compuesta, es una enfermedad que se distribuye principalmente en áreas donde viven personas de raza negra. Tiene una incidencia de uno por 800 aproximadamente entre afroamericanos, del 18% en África y del 1% en Europa, pudiendo presentarse en uno de cada cuatro individuos en las zonas endémicas, en torno a los ríos Níger y Congo. Si consideramos los síndromes drepanocíticos en nuestro país, aparece un caso por cada 100 000 nacidos y en Andalucía menos de cinco por millón; la cuarta parte de ellos serían SC5.
Etiología
La causa se encuentra en una alteración congénita de la hemoglobina. La hemoglobina A1, la más frecuente fuera del periodo fetal/neonatal, consta de dos cadenas alfa y dos cadenas beta, y un total de cuatro grupos hemo con moléculas de hierro que captan el oxígeno. Su conformación espacial tridimensional cambia en función de este hecho, distinguiendo entre hemoglobina oxigenada o desoxigenada. En la hemoglobina S cambia el aminoácido glutámico en la posición 6 de la cadena beta, dando lugar a valina. En el caso de la hemoglobina C, este cambio es por lisina4.
Genética
Para que exista hemoglobina SC debe darse una heterocigosis compuesta de ambos rasgos, en sendos alelos que codifican la cadena beta normal, por lo que no existe hemoglobina A1. Este cambio de aminoácidos se produce por una mutación puntual de nucleótido único en el cromosoma 11. Ambos rasgos siguen un patrón mendeliano de herencia autosómica recesiva por lo que necesariamente un progenitor debe ser portador para HbS y otro para HbC. Por probabilidad, de cada cuatro hijos, un hijo heredaría un rasgo S, un segundo hijo un rasgo C, un tercero de ellos ambos rasgos –SC, como en el caso que nos ocupa–, y un cuarto hijo no heredaría rasgo alguno1.
Fisiopatología
Estos aminoácidos, que tienen carga diferente al original, provocan cambios estructurales que, en el caso de la hemoglobina C, provocan su cristalización con estructura tetragonal u octogonal en función de su estado de oxigenación. Además, se asocia a una alteración en la permeabilidad de membrana al cloro y potasio provocando una corriente de salida que desencadena en una deshidratación del hematíe que adopta morfología de dianocito1,6.
Por otra parte, la hemoglobina S permite la polimerización cuando cambia a forma desoxigenada, formando agregados que precipitan en el interior del hematíe que adopta forma de hoz, obstruyendo la microcirculación y dando lugar a regiones isquémicas.
Hay que considerar que ni el rasgo falciforme ni el cristalizable provocan clínica de forma aislada, además de que ambos fenómenos de polimerización no pueden transcurrir de forma simultánea en un mismo eritrocito puesto que, como hemos explicado, tienen lugar en zonas de la microcirculación con diferente presión parcial de oxígeno. Lo particular de esta enfermedad es que se da una situación paradójica, puesto que la deshidratación del hematíe que acompaña al rasgo para la hemoglobina C es un estímulo independiente para la polimerización de la hemoglobina S y falciformación1.
Clínica
Estos fenómenos combinados tienen una significación clínica que recuerda a una forma atenuada de drepanocitosis clásica:
- Anemia.
- Colelitiasis.
- Fenómenos vasooclusivos (retinianos, infartos óseos, síndrome torácico agudo).
- Esplenomegalia con asplenia funcional.
- Infecciones por gérmenes capsulados.
- Fallo renal.
- Complicaciones obstétricas y perinatales.
- Retraso en el desarrollo.
- Esperanza de vida acortada (≃ alrededor de los 60 años).
A diferencia de la drepanocitosis clásica, presenta una anemia discreta y una esplenomegalia con función conservada durante mucho más tiempo, al igual que en el caso de la función renal. Los cuadros vasooclusivos son mucho menos frecuentes; la retinopatía, al contrario que en la forma homocigota, es proliferativa y como no existe obstrucción completa, la hipoxemia actúa como estímulo para la formación de neovasos, dando lugar a una forma distintiva y de difícil manejo1,6.
Diagnóstico
Una vez planteada la sospecha mediante la clínica y los hallazgos sugestivos de una anemia hemolítica, se confirmaría el diagnóstico mediante un test de falciformación en vacío, la electroforesis de cadenas de hemoglobina, reacción en cadena de la polimerasa del gen alterado o la cromatografía de alta presión, método que puede ser usado como herramienta de cribado neonatal7.
Tratamiento
El manejo se basa en el tratamiento de soporte de la anemia, con administración de folatos –como en todas las hemólisis crónicas con alta proliferación medular, para evitar crisis megaloblásticas– y transfusiones cuando se requieran, nunca superando los 10 mg/dl de hemoglobinemia, puesto que aumentaría la viscosidad y los fenómenos vasooclusivos, siempre con filtros leucocitarios y previa realización de test de Coombs indirecto. El tratamiento etiológico con hidroxiurea no se encuentra estandarizado en niños y el uso de clotrimazol y magnesio, que actuarían evitando la sinergia del mecanismo fisiopatológico por bloqueo directo de la corriente de flujo de potasio, ha demostrado resultados discretos en estudios in vivo8-10.
En cuanto a las inmunizaciones, hay que considerar el estado de asplenia funcional que provoca una susceptibilidad aumentada a la sepsis por gérmenes capsulados6. Además de otras vacunas, los pacientes deben ser vacunados frente al neumococo, Hemophilus influenzae b, meningococo C, hepatitis B, hepatitis A, varicela y gripe. En los pacientes que vayan a viajar a sus países de origen, se deben incluir otras vacunas dependiendo del lugar, como fiebre amarilla, fiebre tifoidea o meningitis A + C. Hay que recordar en estos casos realizar una adecuada profilaxis antimalaria1.
Descompensaciones
Según las guías de práctica clínica vigentes, los pacientes tendrán indicación de acudir a Urgencias si presentan los siguientes síntomas: fiebre, dolor torácico o dificultad respiratoria, dolor óseo o abdominal persistente, síntomas neurológicos o priapismo1.
PROGRAMA DE EDUCACIÓN SANITARIA: ABORDAJE EN ATENCIÓN PRIMARIA
Los pacientes con anemia falciforme requieren una atención integral que aúne el cuidado habitual de cualquier niño en Atención Primaria y los servicios especializados multidisciplinares. El manejo podría agruparse en cuatro grupos etarios1.
Menores de un año
- Al diagnóstico: consejos, información por escrito sobre la enfermedad, resolver dudas. En las primeras visitas no sobrecargar a las familias con muchos detalles. Informar sobre los recursos disponibles en el centro, las asociaciones de pacientes y los programas de apoyo.
- Alimentación: fomentar la lactancia materna y una adecuada hidratación.
- Evitar la guardería.
- Síntomas de alarma del primer año (dolor y síntomas neurológicos son raros a esta edad).
- Repasar las vacunas.
- Evaluar si han entendido el consejo genético y discutir si desean análisis de otros miembros de la familia.
- Adiestramiento para palpar el bazo.
- Evaluación psicosocial.
- Enfatizar la importancia de las revisiones periódicas. Propuesta de temas para las reuniones siguientes.
De uno a cinco años
- Alimentación: si la ingesta de fólico parece baja, recomendar suplementos.
- Recomendar que se respeten los periodos de descanso y sueño.
- Repasar las vacunas.
- Evaluar cómo se enfrentaron a episodios recientes de dolor.
- Aconsejar ejercicio suave-moderado.
- Planear la información a educadores.
Mayores de cinco años
- Informar de los síntomas del priapismo.
- Repasar las vacunas.
- Informar de síntomas neurológicos.
- Colegio: relación con sus compañeros, indagar sobre cambios en rendimiento escolar o comportamiento.
- Evaluación psicosocial.
- A una edad apropiada, empezar a dialogar con el niño sobre la naturaleza de su enfermedad.
Adolescentes
- Dialogar ampliamente sobre la naturaleza de la enfermedad y su impacto en la adolescencia.
- Fomentar la independencia y el autocuidado.
- Revisar el rendimiento escolar y hacer hincapié en síntomas neurológicos.
- Evitar hábitos tóxicos.
- Informar sobre el posible retraso ponderal transitorio, ictericia o cicatrices (cuidado de las úlceras en las piernas).
- Dialogar sobre el priapismo en los varones.
- Evitar inmersiones en agua fría.
- Información sobre sexualidad, herencia de la enfermedad; riesgo de teratogénesis y oligospermia con hidroxiurea.
- Evaluación psicosocial.
RESOLUCIÓN DEL CASO CLÍNICO
El paciente evolucionó favorablemente, con la desaparición de los vómitos, la fiebre y el dolor abdominal, con tratamiento de sostén que incluyó sueroterapia y antitérmicos-analgésicos. Dos meses después de su ingreso presentaba signos de mejoría en el hemograma, no habiendo presentado nuevos episodios relacionados con su enfermedad.
Se realizó además un estudio familiar, que determinó un fenotipo materno C (rasgo cristalizable), un fenotipo paterno S (rasgo falciforme) y encontramos además una drepanocitosis heterocigota combinada en la hermana que tuvo que ser transfundida en Nigeria. Otra de las hermanas presentaba un rasgo C, aunque desconocemos el fenotipo del otro hermano, puesto que no acudió a consulta.
BIBLIOGRAFÍA
- Cantalejo MA. Guía de práctica clínica sobre enfermedad de las células falciformes pediátrica. Sociedad española de hematología y oncología pediátricas 2010. En: Foro Pediátrico de Cartagena [en línea] [consultado el 17/09/2015]. Disponible en: http://www.fpct.es/pdf/DP-SEHOP_2010.pdf
- Martínez-Martínez L, Tovar-Larrucea JA. Dolor abdominal agudo en el niño. En: Cruz M. Manual de Pediatría. 3.ª edición. Madrid: Ergon; 2013. p. 628-30.
- Rives-Solá S. Hemoglobinopatías y síndromes talasémicos. En: Cruz M. Manual de Pediatría. 3.ª edición. Madrid: Ergon; 2013. p. 862-6.
- DeBaun M, Frei-Jones M, Vichinsky E. Hemoglobinopatías. En: Kliegman RM, Arvin AM (eds.). Nelson. Tratado de Pediatría. 19.ª edición. Barcelona: Elsevier; 2012. p. 1662-70.
- Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (Hb S) allele and sickle cell disease: a huge review. Am J Epidemiol. 2000;151:839-45.
- Nagel R, Fabry M, Steinberg M. The paradox of hemoglobin SC disease. Blood Rev. 2003;17:167-78.
- Quinn C. Sickle cell disease in childhood. From newborn screening through transition to adult medical care. Pediatr Clin N Am. 2013;60:1363-81
- Kanter J, Kruse-Jarres R. Management of sickle cell disease from childhood through adulthood. Blood Rev. 2013;27:279-87.
- Sun K, Xia Y. New insights into sickle cell disease: a disease of hipoxia (review). Curr Opin Hematol. 2013;20:215-21.
- Manwani D, Frenette P. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Hematology Am Soc Hematol Educ Program. 2013;2013:362-9.