


Orientación diagnóstica del niño con macro- o microcefalia
MEDIDA DEL PERÍMETRO CEFÁLICO
En primer lugar, para poder obtener un diagnóstico adecuado de macro- o microcefalia es preciso hacer una medida correcta del perímetro cefálico (PC). No son raros los errores en esta sencilla técnica, con las consecuencias que esto puede traer (preocupaciones y pruebas innecesarias). Un error de 0,5 cm puede hacer que el PC cambie de línea de percentil en la curva.
La medida debe tomarse, si es posible, siempre por la misma persona y con la misma cinta métrica. Son preferibles las cintas métricas de tela, que se adaptan mejor al contorno de la cabeza del niño.
Se deben tomar puntos de referencias fijos. Lo más útil es tomar los puntos más prominentes a nivel frontal y occipital. Este último suele ser fácilmente identificable y coincide con la zona central del hueso occipital (zona de crecimiento). Sin embargo, la zona más abultada en la zona frontal no siempre coincide con la glabela, como afirman algunos textos, sino que puede estar por encima de ella, debido a la forma del hueso frontal (Figura 1).
Figura 1. Medida del perímetro cefálico. Mostrar/ocultar
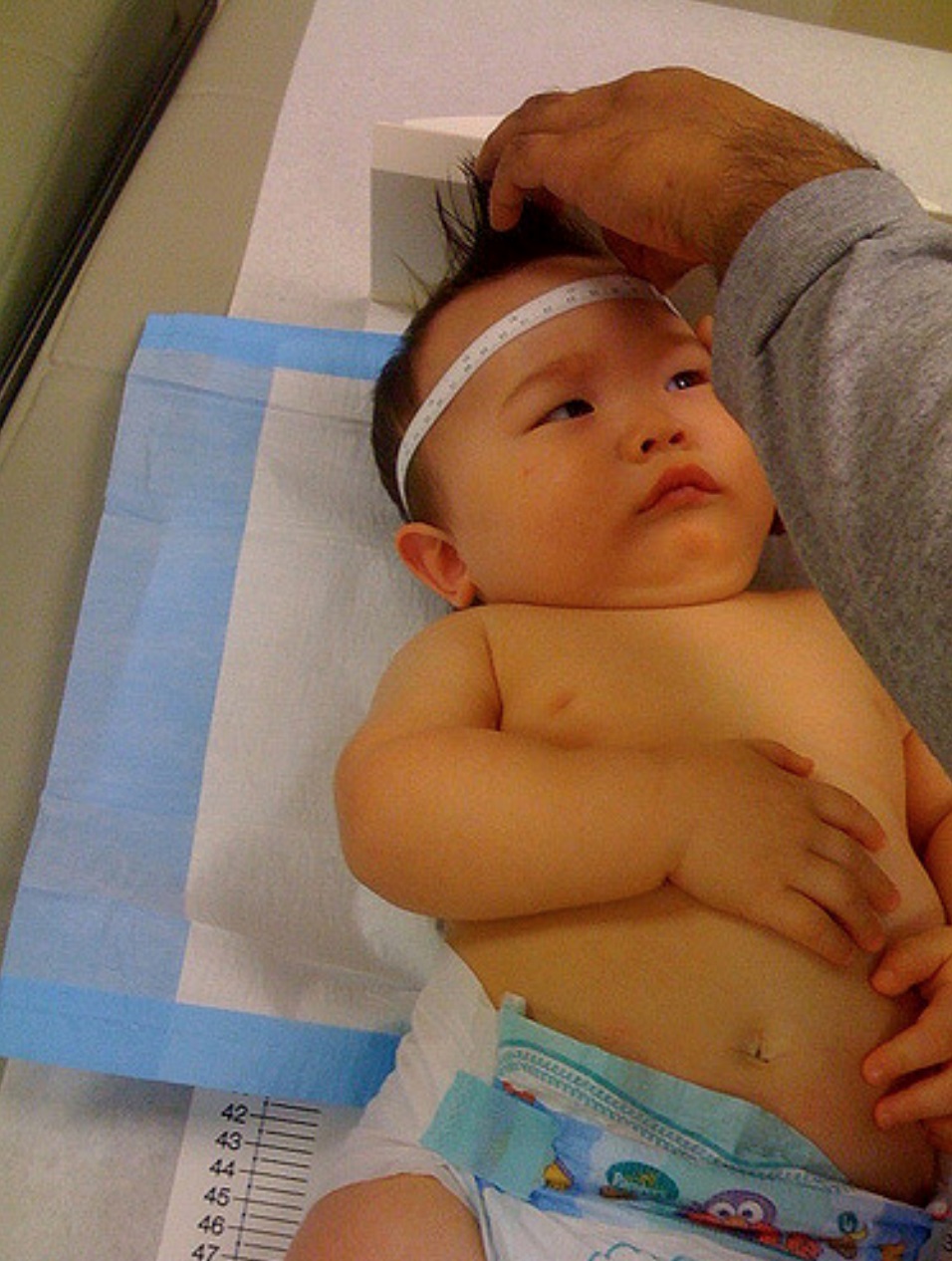
Hay que tener en cuenta también la forma de la cabeza. En los niños con dolicocefalia (diámetro anteroposterior mayor que el transversal) el perímetro craneal suele ser menor, ya que estamos midiendo una elipse y no una circunferencia, a pesar de que el volumen craneal sea normal. Algo similar ocurre en la braquicefalia (diámetro anteroposterior menor que el transversal).
Hay que evitar tomar decisiones con medidas aisladas del PC. Lo importante es la evolución de las medidas en el tiempo, es decir, la velocidad de crecimiento craneal. Si la medida que tomamos es discordante con las previas habría que repetirla hasta 2-3 veces y anotar la media. Si sigue manteniéndose la diferencia, suele ser debido a un error (distintas cintas métricas o persona diferente que la ha realizado). Para asegurarse conviene esperar una semana y volver a medir el PC.
Dado que en nuestro sistema sanitario es práctica habitual la medida del PC junto con la medida de talla y peso en los controles del niño sano, es raro que aparezca en consulta del especialista, de pronto, un niño con una macrocefalia o una microcefalia. Lo habitual es que si esa alteración del tamaño craneal está ya presente en el nacimiento se haya detectado a nivel hospitalario y si se produce a lo largo del periodo de lactante o posterior se detecte a nivel de Atención Primaria. En este último caso, se vería como un aumento o disminución progresivo en la curva de PC.
Los mecanismos genéticos de crecimiento craneal se ponen en marcha durante los 2-3 primeros meses de vida, por lo que los niños que van a ser constitucionalmente macro- o microcéfalos no tienen porqué serlo ya desde el nacimiento, como veremos en los siguientes apartados, sino que pueden nacer con un PC normal y durante los tres primeros meses su curva de crecimiento craneal va ascendiendo o descendiendo progresivamente, saltándose líneas de percentil hasta colocarse por encima del P 97 o por debajo del P 3. Posteriormente la velocidad de crecimiento craneal se estabiliza y el PC se sitúa paralelo a esos percentiles.
La solicitud o no de una ecografía cerebral o la derivación del paciente al neuropediatra va a depender, sobre todo, de que el desarrollo psicomotor y la exploración sean normales (ausencia de rasgos dismórficos, manchas cutáneas y visceromegalias), del PC de los padres y de que el peso y la talla sean concordantes con el PC (un niño normoconfigurado suele tener la talla en percentiles mayores que el peso y el PC en percentiles mayores que la talla).
MACROCEFALIA
Se habla de macrocefalia cuando el PC está por encima del P 97 o de 2 desviaciones estándar (DE) para su edad y sexo.
Aunque, en teoría, la macrocefalia podría ser originada por un aumento de cualquiera de los componentes del cráneo (encéfalo, líquido cefalorraquídeo [LCR], sangre o hueso), en la práctica la mayoría de las macrocefalias son debidas a un aumento de tamaño del encéfalo (megalencefalia) o a un aumento de la cantidad de LCR (hidrocefalia). El aumento del volumen de sangre intracraneal (hemorragia parenquimatosa, hematoma epi- o subdural) o el engrosamiento óseo (talasemia mayor, displasias craneales) son causas muy raras de macrocefalia.
Megalencefalia familiar o macrocefalia familiar benigna
Es la causa más frecuente de macrocefalia en la infancia. Aproximadamente un 2% de la población sana tiene macrocefalia y la mayoría son familiares, por lo que es muy importante valorar siempre el PC de los padres. Suele tener una herencia autosómica dominante.
En estos casos el PC suele ser grande al nacimiento (Figura 2), pero en otras ocasiones puede estar dentro del rango normal y en los 2-3 primeros meses presentan un aumento de la velocidad de crecimiento craneal, saltando líneas de percentil, hasta colocarse por encima del P 97. Después la línea de crecimiento se hace paralela a ese percentil. Esto ocurre antes de los 6 meses1.
Figura 2. Macrocefalia familiar benigna. Mostrar/ocultar
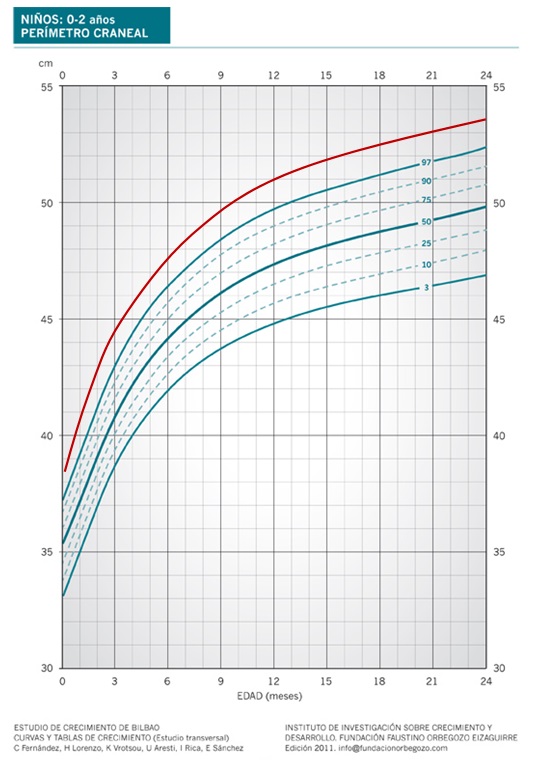
La ecografía cerebral, si se realiza, es normal.
Aumento benigno del espacio subaracnoideo
Es la segunda causa de macrocefalia infantil. Esta denominación es la más adecuada ya que resume las características anatómicas y evolutivas del cuadro. El aumento del PC se hace a expensas de un aumento del espacio subaracnoideo frontal, teniendo el encéfalo un volumen normal. Hay que evitar la denominación de “hidrocefalia externa”, ya que el término de hidrocefalia induce a error y preocupación de los padres.
Al nacimiento el PC puede ser normal, aunque suele estar cerca del P 90, para en los siguientes meses situarse por encima y paralelo al P 97 (Figura 3). Es en estos primeros meses cuando se puede plantear la duda de solicitar una prueba de neuroimagen.
Figura 3. Aumento benigno del espacio subaracnoideo. Mostrar/ocultar
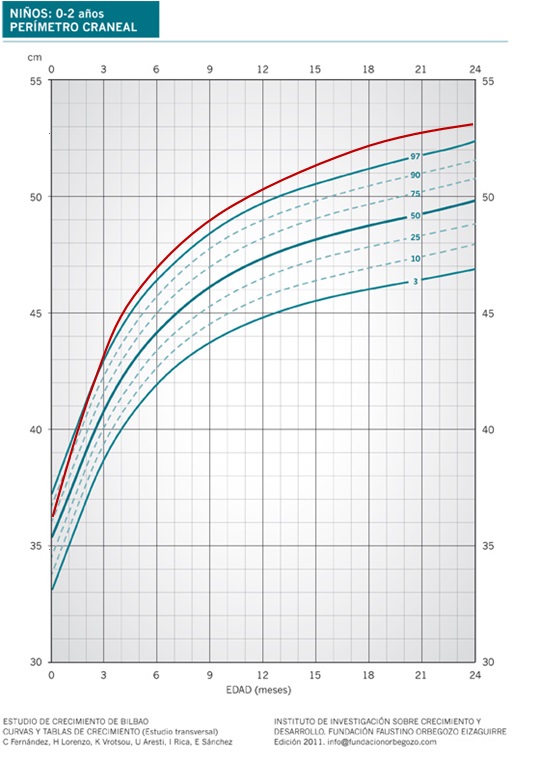
Si se realiza, se observará el aumento del espacio subaracnoideo a nivel frontal. En algunos casos también ampliación de las cisuras de Silvio y de los demás surcos. El tamaño ventricular será normal o mínimamente aumentado, esto lo diferencia de las auténticas hidrocefalias.
La evolución es buena y son lactantes que se desarrollan posteriormente con normalidad, aunque se han descrito con más frecuencia leves problemas de aprendizaje y lectoescritura durante la época escolar2.
Hidrocefalias
La gráfica de PC de un lactante con hidrocefalia sería como la que se muestra en la Figura 4.
Figura 4. Hidrocefalia. Mostrar/ocultar
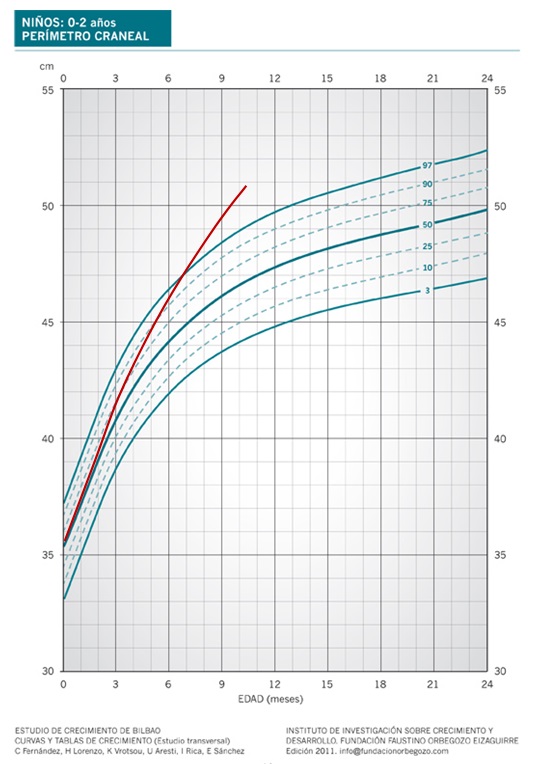
La hidrocefalia es un trastorno en el que el sistema ventricular cerebral contiene una cantidad excesiva de LCR, lo que provoca un aumento de la presión y la dilatación. Puede ser causada por una mayor producción, disminución de la absorción u obstrucción del flujo de LCR.
El mielomeningocele es la causa más frecuente de hidrocefalia congénita y ocurre en el 15-20% de los casos3,4. La causa más frecuente de hidrocefalia adquirida es la secundaria a hemorragias intraventriculares, típica de los prematuros5.
En estos casos siempre está indicada la prueba de imagen, que habitualmente suele ser una ecografía cerebral por su rapidez y facilidad de realización. Posteriormente si queremos completar el estudio se realizará una resonancia magnética (RM) craneal, intentando evitar la tomografía computarizada (TC) para no radiar al paciente.
La ecografía puede mostrarnos un aumento de los ventrículos con espacios subaracnoideos normales, hablamos entonces de una hidrocefalia no comunicante, cuya causa más frecuente es la estenosis congénita del acueducto de Silvio, o bien, unos ventrículos y espacios subaracnoideos aumentados. Se trata entonces de una hidrocefalia comunicante y la causa más frecuente suele ser la hemorragia intraventricular perinatal.
Mielomeningocele
El 90% de los mielomeningoceles tienen algún grado de dilatación ventricular, el que desarrollen hidrocefalia franca depende de diversos factores, entre ellos la altura del defecto raquídeo. La hidrocefalia se debe a la malformación de Chiari II.
Hidrocefalia poshemorragia intraventricular perinatal
Aunque un 40% de estas hidrocefalias se estabilizan en el periodo neonatal, puede ocurrir también una progresión lenta que se manifieste como macrocefalia progresiva durante los primeros meses de vida6.
Estenosis congénita del acueducto de Silvio
Supone el 11% de las hidrocefalias en lactantes. Puede ser debida a un defecto genético (gen L1 en Xq28)7, secundaria a la inflamación por una infección intrauterina o formar parte de otras patologías como la neurofibromatosis tipo I.
Otras causas de hidrocefalia
- Tumores cerebrales congénitos.
- Síndromes de Klippel-Feil o de Walker-Warburg, acondroplasia.
- Malformaciones: Dandy-Walker, aneurisma de la vena de Galeno.
- Abscesos, hematomas o meningitis.
Tratamiento
En el caso de las hidrocefalias, drenaje o derivación ventriculoperitoneal y tratamiento de la causa subyacente. Seguimiento clínico si se trata de un aumento benigno del espacio subaracnoideo.
MICROCEFALIA
Se considera microcefalia si el PC se encuentra por debajo de -2 DE o del P 3 para la edad y sexo del paciente. Hay que tener en cuenta también la etnia del sujeto. Conviene corregir la edad según la edad gestacional en el caso de los niños con antecedentes de prematuridad, sobre todo por cuestiones prácticas a la hora de reflejar la evolución de los sucesivos PC en la gráfica.
Por definición, un 2-3% de la población es microcéfala y la inmensa mayoría no tendrán problemas neurológicos posteriores. Pero si el PC se encuentra por debajo de -3 DE hablamos de microcefalia grave y las posibilidades de problemas neurológicos posteriores (discapacidad cognitiva, epilepsia o parálisis cerebral infantil [PCI]) son aproximadamente de un 80%8.
Existen casos en los que no se cumple el criterio de microcefalia (PC
Hasta en un 41% de los casos de microcefalia no se encuentra la causa9.
Patogenia
Debido a que el crecimiento del encéfalo induce el crecimiento el cráneo, si no hay crecimiento encefálico se producirá habitualmente una microcefalia, aunque puede haber también encéfalos pequeños en cráneos normales.
Existen dos mecanismos principales:
- Falta de desarrollo cerebral o desarrollo cerebral anormal relacionado con un insulto importante durante el periodo específico de inducción y migración celular; este tipo de microcefalia se cree que es el resultado de una reducción en el número de neuronas generadas durante la neurogénesis; el cerebro anterior está más gravemente afectado (por ejemplo, holoprosencefalia).
- Lesión o insulto a un cerebro previamente normal (a veces llamado microcefalia secundaria); este tipo de microcefalia se cree que es el resultado de una reducción en el número de procesos dendríticos y conexiones sinápticas.
Clasificación
La clasificación más adecuada de la microcefalia es la que se realiza atendiendo a si está presente en el momento del nacimiento (congénita) o aparece posteriormente (posnatal).
Realmente esta división no ayuda mucho de cara al estudio diagnóstico, ya que una misma causa puede dar una microcefalia congénita o adquirida dependiendo del momento en que actúe sobre el encéfalo.
Microcefalia congénita
Algunos autores la denominan también microcefalia primaria, pero hay que intentar evitar este término ya que lleva a la confusión con la microcefalia vera, también denominada a veces microcefalia primaria.
La causas principales son infecciones intrauterinas, tóxicos prenatales, anomalías cromosómicas, síndromes polimalformativos y accidentes vasculares prenatales.
En estos casos el estudio se realiza habitualmente en el periodo neonatal y suele consistir en una prueba de neuroimagen (habitualmente RM), estudio genético (si hay rasgos dismórficos asociados), serología TORCH y estudio metabólico.
Microcefalia vera
Se la denomina también microcefalia primaria, microcefalia verdadera o MCPH. Es un raro trastorno autosómico recesivo, genéticamente heterogéneo, que se caracteriza por una microcefalia (habitualmente PC <3 DE) que está presente en el momento del nacimiento (Figura 5) y en la que no suele haber alteraciones anatómicas en el encéfalo. Estos niños no tienen sintomatología neurológica perinatal y posteriormente desarrollan trastornos del aprendizaje habitualmente leves o moderados, aunque en algunos casos pueden ser graves. Un 50% de los casos son debidos a una mutación en el gen ASPM (1q31)10.
Figura 5. Microcefalia vera. Mostrar/ocultar
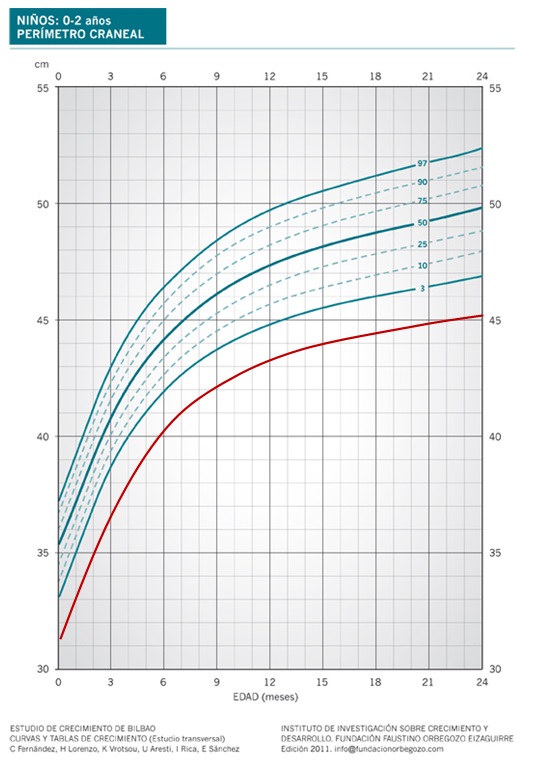
Microcefalia por infecciones prenatales
La infección prenatal causante de microcefalia congénita más frecuente es la producida por citomegalovirus (CMV). Puede llegar a manifestarse en el momento del parto solamente con microcefalia, sin ningún otro dato clínico añadido. Por ello es importante la determinación de anticuerpos y reacción en cadena de la polimerasa (PCR) lo antes posible después del nacimiento, ya que si retrasamos la determinación podemos obtener una positividad, pero debida a infección posnatal. En los casos en que han pasado las primeras semanas o meses se puede recurrir a realizar la PCR para CMV en la sangre recogida en papel para el cribado neonatal.
Una causa infecciosa frecuente de microcefalia en países sudamericanos es la infección prenatal por virus Zika.
Ingesta de tóxicos o malnutrición en la madre
En estos casos la microcefalia suele formar parte de un cuadro de crecimiento intrauterino retardado (CIR). Destaca el síndrome alcohólico-fetal. Las consecuencias de la exposición del feto al alcohol incluyen retraso del crecimiento pre- y posnatal, fisuras palpebrales cortas, surco plano y labio superior delgado.
Anormalidades neuroanatómicas
Las anomalías neuroanatómicas que se asocian con la microcefalia incluyen defectos del tubo neural, holoprosencefalia, atelencefalia, lisencefalia, esquizencefalia, polimicrogiria, macrogiria y la secuencia de disrupción cerebral fetal.
En el caso de la holoprosencefalia suele ir acompañada de alteraciones en la formación de la línea media facial (hipotelorismo, incisivo único, cebocefalia, ciclopia), lo que hace más fácil el diagnóstico. En la atelencefalia la microcefalia es muy llamativa. La lisencefalia puede ser de origen genético o infeccioso y la microcefalia suele estar ya presente en el momento del nacimiento.
Microcefalia posnatal
También denominada secundaria. El niño nace con un PC normal y en los meses siguientes va desarrollando progresivamente una microcefalia (Figura 6). La etiología es múltiple y puede ser causada por todos aquellos procesos que afecten de forma importante al encéfalo durante el periodo pre-, peri- o posnatal. Pueden ser enfermedades metabólicas, heredodegenerativas, malnutrición, pero sobre todo la encefalopatía hipóxico-isquémica.
Figura 6. Microcefalia familiar. Mostrar/ocultar
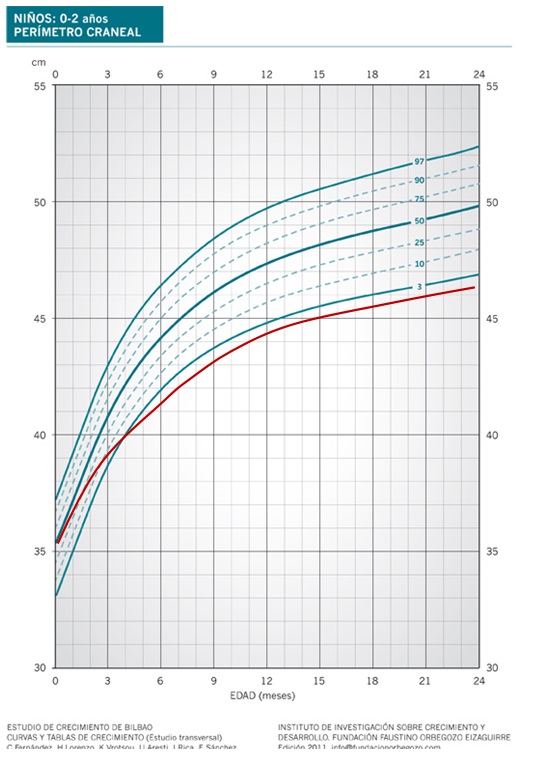
Encefalopatía hipóxico-isquémica
El desarrollo de una microcefalia en un niño con historia de encefalopatía hipóxico-isquémica perinatal va a implicar un pronóstico severo con discapacidad cognitiva importante, epilepsia y PCI (Figura 7).
Figura 7. Craneosinostosis secundaria a encefalopatía hipóxico-isquémica grave. Mostrar/ocultar
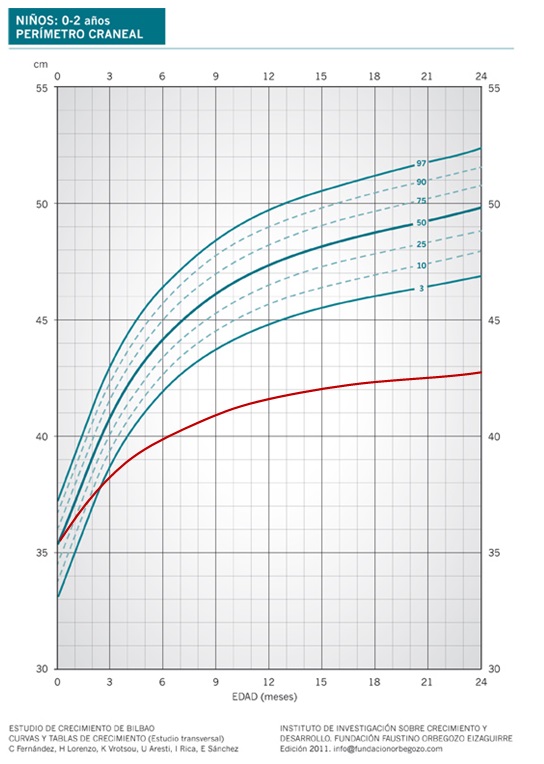
Malnutrición y enfermedades crónicas posnatales
En estos niños todo el crecimiento se retrasa. Se afectará primero el peso, posteriormente la talla y por último el PC.
Lesiones cerebrales posnatales
Infartos cerebrales, encefalitis y daños cerebrales secundarios a malos tratos.
Enfermedades metabólicas
Los trastornos metabólicos rara vez se presentan con microcefalia.
Craneosinostosis
El cierre precoz de todas las suturas craneales es muy raro y cuando aparece suele ser secundario a una atrofia cerebral importante, como ocurre en las encefalopatías hipóxico-isquémicas graves. En estos casos al palpar el cráneo se aprecian crestas a nivel de todas las suturas.
En los casos de craneosinostosis primarias suele afectarse solo una sutura, palpándose una cresta a ese nivel y siempre se acompañan de deformidad craneal, pero no de microcefalia. La craneosinostosis más frecuente es el cierre precoz de la sutura sagital, que produce como consecuencia una dolicocefalia/escafocefalia.
Las plagiocefalias y braquicefalias posturales son muy frecuentes, pero no son causa de microcefalia.
Diagnóstico
Como en el caso de las macrocefalias, el estudio principal es la neuroimagen. El problema que se plantea en estos casos es que muchas veces el tamaño mínimo de la fontanela anterior no permite la realización de una ecografía cerebral, por lo que hay que recurrir a la RM craneal.
Si la microcefalia está presente al nacimiento, habrá que intentar descartar por la historia clínica la ingesta de alcohol o tóxicos por la madre, malnutrición, traumatismos durante el embarazo, episodios febriles, etc. Es recomendable la realización de serología TORCH y estudio genético.
Tratamiento
El tratamiento es el de la causa de la microcefalia: antibióticos o antivirales en el caso de infecciones pre- o perinatales del sistema nervioso central, tratamiento de la encefalopatía hipóxico-isquémica (preventivas, hipotermia) y restricción del sustrato tóxico y aporte de cofactores y vitaminas en las enfermedades metabólicas. Cirugía correctora en el caso de craneosinostosis primaria.
BIBLIOGRAFÍA
- Lorber J, Priestley BL. Children with large heads: a practical approach to diagnosis in 557 children, with special reference to 109 children with megalencephaly. Dev Med Child Neurol. 1981;23:494-504.
- Álvarez LA, Maytal J, Shinnar S. Idiopathic external hydrocephalus: natural history and relationship to benign familial macrocephaly. Pediatrics. 1986;77:901-7.
- Jeng S, Gupta N, Wrensch M, Zhao S, Wu YW. Prevalence of congenital hydrocephalus in California, 1991-2000. Pediatr Neurol. 2011;45:67-71.
- Persson EK, Anderson S, Wiklund LM, Uvebrant P. Hydrocephalus in children born in 1999-2002: epidemiology, outcome and ophthalmological findings. Childs Nerv Syst. 2007;23:1111-8.
- Tully HM, Dobyns WB. Infantile hydrocephalus: a review of epidemiology, classification and causes. Eur J Med Genet. 2014;57:359-68.
- Whitelaw A, Aquilina K. Management of posthaemorrhagic ventricular dilatation. Arch Dis Child Fetal Neonatal Ed. 2012;97:F229-3.
- Schrander-Stumpel C, Fryns JP. Congenital hydrocephalus: nosology and guidelines for clinical approach and genetic counselling. Eur J Pediatr. 1998;157:355-62.
- Ashwal S, Michelson D, Plawner L, Dobyns WB. Practice parameter: evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2009;73:887-97.
- Von der Hagen M, Pivarcsi M, Liebe J, von Bernuth H, Didonato N, et al. Diagnostic approach to microcephaly in childhood: a two-center study and review of the literature. Dev Med Child Neurol. 2014;56:732-41.
- Létard P, Drunat S, Vial Y, Duerinckx S, Ernault A, Amram D, et al. Autosomal recessive primary microcephaly due to ASPM mutations: an update. Hum Mutat. 2018;39:319-32.
LECTURAS RECOMENDADAS
- Arzimanoglou A. Aicardi’s diseases of the nervous system in childhood. MacKeith Press; 2018. p. 1524.
- Piña-Garza E. Fenichel’s clinical pediatric neurology. Filadelfia: Elsevier Saunders; 2013. p. 412.
- Verdú A. Manual de Neurología Infantil. Madrid: Editorial Panamericana; 2014. p. 1350.