

Tristeza en el niño como síntoma guía de patología orgánica
2 MIR-Pediatría. Complejo Asistencial Universitario de León. León. (España).
3 Servicio de Pediatría. Complejo Asistencial Universitario de León. León. (España).
CASO CLÍNICO
Motivo de consulta: niña de 5 años que acude a consulta de Atención Primaria acompañada de su madre, quien refiere cambio conductual en la paciente a lo largo de los últimos 8 meses.
Antecedentes familiares: sin interés.
Antecedentes personales: enfermedad celíaca diagnosticada a los 2 años, en tratamiento con dieta exenta de gluten. Desarrollo psicomotor y ponderoestatural adecuados. Correctamente vacunada. No tiene alergias a medicamentos conocidas.
Enfermedad actual: la madre refiere que, en los 8 meses previos a la consulta, encuentra a la paciente triste, con tendencia al mutismo, quejas frecuentes de diferentes dolencias (cefalea, mareos, dolor abdominal…) y apatía ante actividades que previamente solía disfrutar, como ir a la playa. Coincide en el tiempo con el embarazo de una tía y el traslado a otro domicilio. Niega cambios en la estructura familiar o mal ambiente en el ámbito escolar. Presenta un buen control de la enfermedad celíaca.
Exploración física: en consulta la exploración física es normal. No se observan signos de focalidad neurológica. Presenta un patrón de crecimiento sin desviaciones, con peso y talla en el percentil 25.
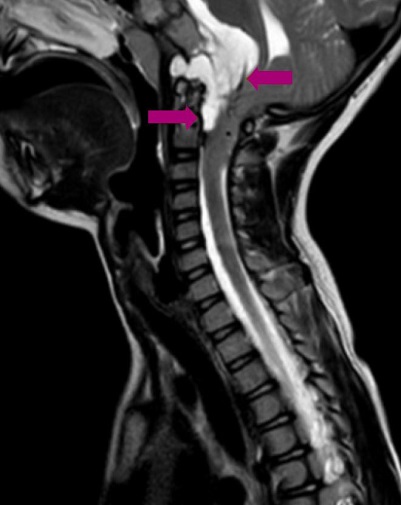
Seguimiento y pruebas complementarias: ante la sospecha de trastorno psicosomático, se deriva a Psiquiatría Infanto-Juvenil, que inicia intervención con apoyo del profesorado. Consulta a los 2 meses por presentar una mayor inhibición del lenguaje. El equipo psicopedagógico indica una valoración otorrinolaringológica para descartar alteraciones que pudieran justificarla. La exploración y la radiografía de adenoides descartan hipertrofia adenoidea u otras alteraciones a ese nivel. La paciente refiere que no ve bien en clase, por lo que se deriva a Oftalmología, donde se diagnostica hipermetropía y estrabismo, por lo que se pautan lentes correctoras y parches de penalización para la corrección de la ambliopía. A los 2 meses de la primera consulta en Oftalmología aparece de forma súbita un estrabismo convergente marcado, por lo que es valorada de nuevo y se deriva al hospital para un estudio neurológico. En la exploración neurológica se observa una paresia del VI par craneal bilateral, marcha inestable con lateropulsión evidente bilateral, maniobra índice-nariz alterada por dismetría bilateral y Romberg positivo. La tomografía computarizada (TC) realizada se informa como normal. Dada la forma de presentación del estrabismo y la sospecha de afectación neurológica, ingresa para realización de resonancia magnética craneal. Se visualiza un tumor localizado en la base del cráneo (Figuras 1 y 2), compatible con un cordoma de clivus.
Figura 1. Tumor en la base del cráneo compatible con un cordoma de clivus. Mostrar/ocultar
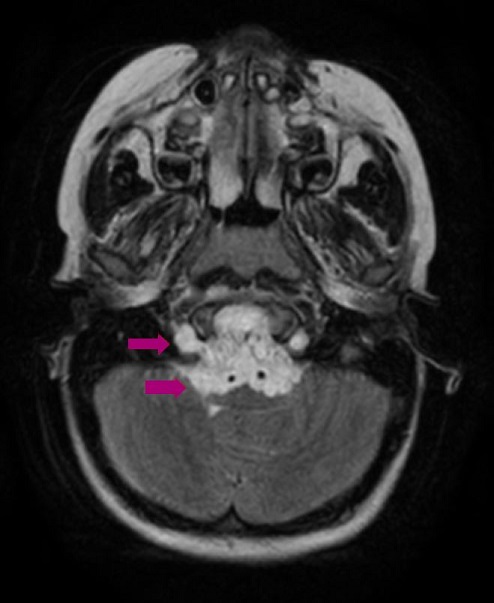
Figura 2. Tumor en la base del cráneo compatible con un cordoma de clivus. Mostrar/ocultar
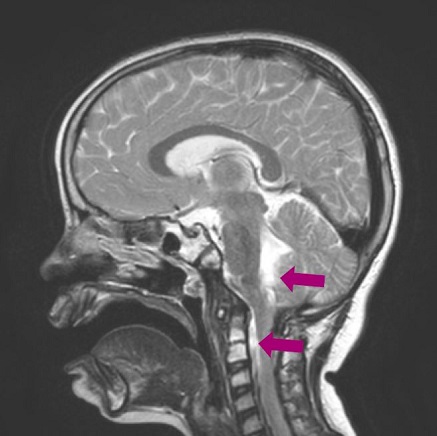
Tratamiento: es intervenida quirúrgicamente, a los 6 años y 6 meses de edad, mediante abordaje endonasal, con resección macroscópicamente completa. A los 4 meses desarrolla telarquia, por lo que se realiza test de hormona adrenocorticotropa (ACTH) y hormona liberadora de la hormona luteinizante (LHRH), con diagnóstico de pubertad precoz central; el resto de la analítica hormonal y la edad ósea se encuentran dentro de los valores de normalidad. Se pauta tratamiento con triptorelina, un análogo de la hormona liberadora de gonadotropinas (GnRH), para frenar la pubertad. A los 4 meses de la intervención se realiza una resonancia magnética de control en la que se observan pequeños restos tumorales a nivel de los agujeros intervertebrales C1-C2. Se recomienda iniciar protonterapia, un tratamiento no disponible actualmente en España, por lo que la paciente se desplaza a Italia para recibirlo durante 2 meses. Tres meses después, en una resonancia magnética de control, se observa tejido que sugiere resto de tumor en la fosa posterior y el espacio epidural anterior a nivel C1-C2 (Figuras 3 y 4), aparentemente estable.
Figura 3. Resto de tumor en la fosa posterior y el espacio epidural anterior a nivel C1-C2. Mostrar/ocultar
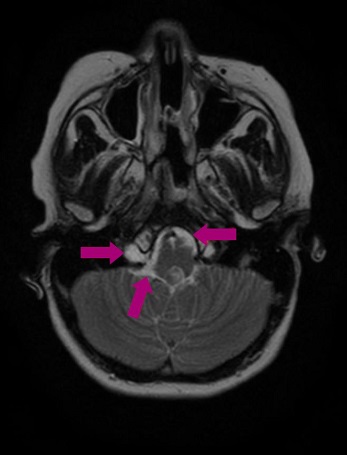
Figura 4. Resto de tumor en la fosa posterior y el espacio epidural anterior a nivel C1-C2. Mostrar/ocultar
Actualmente, con 8 años, mantiene el tratamiento con triptorelina para frenado puberal. En la exploración se observa escaso tejido granular residual en mamas, Tanner 1 (T1, P1, A1). A nivel oftalmológico persiste el estrabismo, por lo que recibe inyecciones periódicas de toxina botulínica. Está contenta y ha finalizado el curso escolar con apoyo pedagógico.
DISCUSIÓN
Los cambios conductuales en la edad pediátrica obedecen a causas muy diversas, tanto de origen psicosomático (depresión, ansiedad, maltrato…) como de origen orgánico (por ejemplo, trastornos digestivos o tumores). No siempre es fácil el diagnóstico precoz, dada la evolución tórpida de muchos de estos procesos. El caso que se presenta refleja esta realidad, ya que, inicialmente, la paciente presenta alteración del ánimo y la conducta, así como quejas frecuentes de diferentes síntomas inespecíficos que, en conjunto, orienta a un origen psicosomático. Con el tiempo, la paciente desarrolla síntomas y signos sugestivos de alteración de la visión, aparece posteriormente un estrabismo muy marcado, dato que motiva la indicación del estudio neurológico.
La resonancia magnética craneomedular muestra una masa en fosa posterior compatible con cordoma de clivus, que es un tumor derivado de restos persistentes de la notocorda. Esta estructura mesodérmica embrionaria da lugar al esqueleto axial y, en el adulto, su presencia se limita a los núcleos pulposos de los discos intervertebrales y puede permanecer en la columna vertebral o la base del cráneo.
Los cordomas suelen aparecer en adultos de entre 40 y 70 años y solo el 5% de los casos se dan en niños. Se localizan con mayor frecuencia a nivel sacro (50%), seguido de la base del cráneo (30%) y el resto a nivel de la columna vertebral (cervical: 10%, torácica: 5%)1,2. La forma de presentación más frecuente (71%) es la alteración de los pares craneales, el VI par es el más frecuentemente afectado (31-57% de los casos) y los síntomas más frecuentes son la cefalea y la diplopía. Sin tratamiento, la supervivencia de los pacientes se encuentra entre los 6 y los 24 meses. El tratamiento de elección en este tipo de tumor se basa en una cirugía radical3,4, lo más extensa posible, teniendo en cuenta la dificultad de resección en las áreas a las que afecta. Si la resección es incompleta, el tumor es de alto grado o se constata recidiva tumoral, se debe asociar radioterapia4. Actualmente se tiende a utilizar radioterapia con partículas (iones de carbono o radiación con protones), de elección frente a la radioterapia convencional por su mayor precisión, por no presentar dosis de salida y porque puede adaptarse a volúmenes irregulares, permitiendo una menor irradiación de los tejidos sanos. Parece que los tumores pediátricos son una de las indicaciones más evidentes de la protonterapia. La creación de protocolos formalizados y de investigaciones coordinadas entre los Servicios de Oncología Pediátrica respecto a la protonterapia serán indispensables para el desarrollo y la evaluación de esta técnica en Pediatría5. Actualmente no se encuentra disponible en España, aunque podría empezar a utilizarse a partir de 2019.
El pronóstico de los pacientes depende del éxito de la cirugía. El cordoma tiene tendencia a la recidiva local y esta es la principal causa de mortalidad; son raras las metástasis a distancia. La supervivencia a los 5 años es del 51-87%, mientras que la supervivencia libre de enfermedad se sitúa en 3,8 años si la exéresis es completa, de 2,1 años si la exéresis es parcial y se asocia radioterapia, y de 8 meses si se realiza resección subtotal sin radioterapia6.
BIBLIOGRAFÍA
- Martínez FJ, Conde E, Manjón P, Ricoy JR, Pérez A. Cordoma. Sus variantes y diagnóstico diferencial. Rev Esp Patol. 2007;40:135-45.
- Erdem E, Angtuaco EC, Van Hemert R, Park JS, Al-Mefty O. Comprehensive review of intracranial chordoma. Radiographics. 2003;23:995-1009.
- Stacchiotti S, Sommer J, Chordoma Global Consensus Group. Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol. 2015;16:71-83.
- Llorente JL, Obeso S, Rial JC, Sánchez-Fernández R, Suárez C. Tratamiento de los cordomas de clivus. Acta Otorrinolaringol Esp. 2010;61:135-44.
- Conde Moreno AJ, Feuvret L, Noel G, Calugaru V, Ferrand R, Delacroix S, et al. La protonterapia: indicaciones y perspectivas. Rev Oncol. 2004;6:403-14.
- Lanzino G, Dumont AS, Lopes MB, Laws ER. Skull base chordomas: overview of disease, management options and outcome. Neurosurg Focus. 2001;10:E12.