


Abordaje de la adrenarquia prematura en Atención Primaria
PUNTOS CLAVE
- La adrenarquia prematura es la aparición de vello púbico o axilar u olor apocrino antes de los 8 años en las niñas y de los 9 años en los niños. Es un motivo de consulta frecuente en Atención Primaria.
- El diagnóstico de adrenarquia requiere clínica compatible y demostración bioquímica de elevación de marcadores androgénicos como dehidroepiandrosterona en su forma sulfato conjugada (DHEA-S).
- La adrenarquia prematura idiopática es la causa más frecuente de hiperandrogenismo en niños prepuberales y su diagnóstico es de exclusión. Es considerada una variante de la normalidad, que no precisa tratamiento, pero se asocia a riesgo moderado de padecer síndrome de ovario poliquístico (SOP), insulinorresistencia, síndrome metabólico del adulto y riesgo cardiovascular. Se recomienda seguimiento de estos pacientes hasta el final de la pubertad.
- Si los niveles de testosterona y DHEA-S están muy elevados en niños con adrenarquia prematura, si la edad ósea está adelantada comprometiendo la talla adulta final, si hay algún hallazgo atípico en la evaluación inicial o existe empeoramiento clínico durante el seguimiento del paciente, es necesario la exclusión de hiperplasia suprarrenal congénita (HSC) no clásica u otros desórdenes virilizantes.
INTRODUCCIÓN
Adrenarquia se refiere a la aparición de signos clínicos como vello púbico (pubarquia) o axilar (axilarquia) o incremento del olor corporal apocrino. Se considera prematura si sucede antes de los 8 años en niñas y de los 9 años en niños. Estos signos físicos son secundarios a un aumento de los precursores adrenales androgénicos en la infancia, principalmente DHEA-S, tras un cambio madurativo funcional de la corteza suprarrenal.
La adrenarquia prematura es un motivo de consulta frecuente en las consultas de Pediatría, pero se desconoce exactamente la prevalencia en la población general. Se estima una ratio de 9 niñas por cada niño varón. La mayoría de los casos son benignos y autolimitados, seguidos de una pubertad normal. Es más frecuente en niñas de raza negra, en obesos e insulinorresistencias.
El pediatra de Atención Primaria debe estar familiarizado con el manejo de la adrenarquia prematura, realizando un enfoque inicial correcto para evitar derivaciones innecesarias a Endocrinología Pediátrica y ser capaz de detectar con rapidez trastornos que indiquen un estudio más extenso o tratamiento.
RECUERDO ANATÓMICO-FISIOLÓGICO
En la glándula suprarrenal se diferencian dos zonas anatómicas: la médula adrenal que segrega catecolaminas (adrenalina, noradrenalina, dopamina) y la corteza suprarrenal, que a su vez se divide en tres zonas concéntricas bien diferenciadas:
- Zona glomerular donde se sintetizan mineralocorticoides, sobre todo aldosterona.
- Zona fascicular, segregando mayoritariamente glucocorticoides en forma de cortisol.
- Zona reticular: es la parte más interna donde se sintetizan los esteroides sexuales.
Durante la vida fetal, la glándula adrenal es un órgano funcionalmente activo produciendo niveles de DHEA-S elevados, pero tras el nacimiento la zona reticular involuciona, disminuyendo así los niveles de androstendiona y de DHEA. Posteriormente, a partir de los 5-6 años existe un cambio morfológico y funcional de la zona reticular que da lugar a la adrenarquia con liberación de andrógenos como DHEA-S y 17-OH-progesterona.
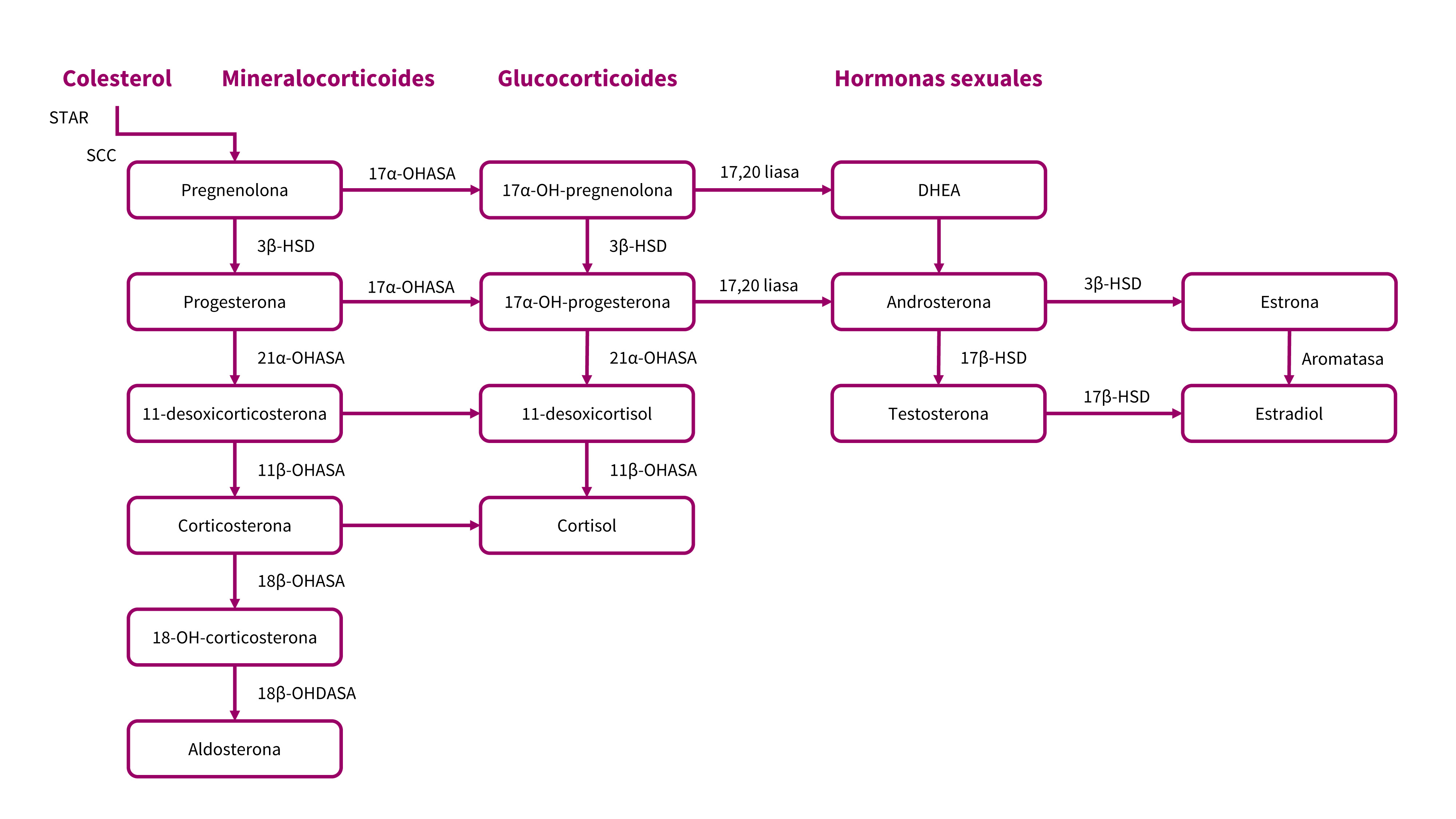
Conviene repasar la esteroidogénesis pues la deficiencia de las enzimas implicadas puede suponer la aparición de un hiperandrogenismo por acúmulo de metabolitos (Figura 1).
Figura 1. Esteroidogénesis suprarrenal. Mostrar/ocultar
ETIOPATOGENIA
Adrenarquia prematura idiopática (API)
Es la causa más frecuente de adrenarquia prematura y se considera una variante de la normalidad. Existe una presentación clínica más marcada, que se conoce como “exagerada”. Se asocia con riesgo de padecer SOP, insulinorresistencia y síndrome metabólico del adulto. La etiología de la API es desconocida; aunque no se han establecido bases genéticas convincentes, se han descrito polimorfismos de genes involucrados en el sistema del factor de crecimiento insulínico, relacionados con insulina o receptor de andrógenos. Por otro lado, el sobrepeso, la obesidad, el hiperinsulinismo, pequeño para la edad gestacional (PEG) y la prematuridad se han descrito como factores predisponentes. Se postula como un evento clave desde el punto de vista molecular la fosforilación del CYP17A1, aumentando su actividad liasa y favoreciendo así la producción de andrógenos adrenales. Es posible que muchos de los factores ambientales, nutricionales u hormonales implicados participen en este incremento enzimático. También se ha descrito mayor sensibilidad de los tejidos periféricos a la acción androgénica.
Trastornos virilizantes
Son menos frecuentes pero su naturaleza progresiva requiere especial vigilancia.
Los trastornos virilizantes puros-francos (por tumores ováricos o adrenales) se sospechan por el rápido desarrollo de vello sexual, acné que requiere tratamiento farmacológico, aumento de musculatura, voz grave y, en chicas, clitoromegalia. Las concentraciones séricas de testosterona y DHEAS están muy por encima del rango normal. Este exceso androgénico produce adelanto significativo de la edad ósea.
Los trastornos virilizantes leves-moderados cursan con progresión de pubarquia, crecimiento y edad ósea de forma más lenta y las concentraciones de DHEAS están en el rango alto de la normalidad. Dentro de estos destaca por su frecuencia la hiperplasia suprarrenal congénita (HSC).
Hiperplasia suprarrenal congénita, forma no clásica o leve
Es un trastorno hereditario de la esteroidogénesis suprarrenal de cortisol, de diagnóstico tardío, ya que el déficit enzimático es parcial. Se debe a mutaciones que producen hiperandrogenismo moderado en forma de adrenarquia prematura (sobre todo pubarquia). No presentan ambigüedad genital ni existe riesgo de insuficiencia adrenal precoz.
Se conocen siete formas clínicas en función del déficit enzimático con diversos grados de afectación:
- Deficiencia de 21-hidroxilasa (CYP21A2): es la causa más frecuente de HSC no clásica.
- Deficiencia de 11-β-hidroxilasa (CYP11B1): clásicamente suele cursar con hipertensión y virilización (incluyendo genitales ambiguos en niñas), pero ocasionalmente se presenta como forma no clásica con hiperandrogenismo moderado.
- Deficiencia de 3-β-hidroxiesteroide deshidrogenasa (HSD3B2): la forma clásica conlleva ambigüedad genital en ambos sexos y síntomas de deficiencia de aldosterona, pero en ocasiones tiene una forma no clásica de presentación tardía con síntomas más leves, como adrenarquia prematura.
- Deficiencia de 17-α-hidroxilasa, StAR, colesterol desmolasa y P450 óxido-reductasa.
Solo mencionar la forma clásica o grave, de diagnóstico neonatal, ante la presencia de ambigüedad genital en niñas o macrogenitosomía en niños. También llamada “pierde sal”, en ausencia de tratamiento produce una crisis adrenal grave con vómitos, deshidratación hiponatrémica, hipotensión y shock a los 7-14 días de vida. Esta forma no plantea un diagnóstico diferencial con la adrenarquia prematura.
Hiperandrogenismo adrenal congénito ACTH dependiente
- Resistencia a glucocorticoides por defectos en la señal del receptor: es una forma congénita que se produce por un feedback negativo del cortisol, inadecuado con el consecuente exceso de hormona adrenocorticotropa (ACTH).
- Deficiencia de cortisona reductasa: defecto en el metabolismo periférico del cortisol que produce un aumento compensatorio de ACTH y un consecuente hiperandrogenismo moderado. Este trastorno raro está producido por deficiencia en la 11-β-hidroxiesteroide deshidrogenasa tipo 1 (HSD11B1) que es la mayor reductasa de cortisona o producido por deficiencia de la hexosa-6-fosfatodeshidrogenasa (H6PDH) que es un cofactor para la cortisona reductasa.
Síndrome de Cushing
Varias formas de síndrome de Cushing debidas a hiperplasia adrenal se asocian en ocasiones a virilización infantil. Algunos casos son por formas hereditarias de hiperplasia adrenocortical y otros por trastornos dependientes de ACTH de inicio tardío.
Iatrogénica, por exposición exógena a andrógenos
Se han descrito casos de pubarquia prematura secundaria al contacto pasivo con los cuidadores que utilizan preparaciones tópicas de andrógenos como terapia hormonal sustitutiva o como ayuda para la función sexual o muscular. Los anabolizantes esteroideos también pueden producir pubarquia prematura y clitoromegalia.
Otros
- Deficiencia aparente de DHEA sulfotransferasa: es debida a un defecto genético en la sulfatación de DHEA, en la que los niveles de DHEAS son indetectables o muy bajos. No se afecta la secreción de corticoides. Es causa de displasia espondiloepimetafisaria y de varios grados de hiperandrogenismo, sobre todo pubarquia prematura.
- Trastorno metabólico androgénico periférico: se han descrito casos de pubarquia prematura en los shunt portosistémicos congénitos.
DIAGNÓSTICO DIFERENCIAL
- Hipertricosis: es el exceso de vello corporal generalizado (tanto en zonas androgénicas como no). Suele tener asociación familiar o estar causada por desórdenes metabólicos (anorexia nerviosa, alteraciones tiroideas) o fármacos (ciclosporina), sin que exista exceso de andrógenos.
- Pubarquia prematura idiopática: niños con pubarquia prematura sin evidencia bioquímica de adrenarquia, es decir, andrógenos séricos están en niveles prepuberales.
- Pubertad precoz: es la presencia de gonadarquia en niños antes de los 9 años (aumento del tamaño testicular >4 cc) y antes de los 8 años en niñas (aparición del botón mamario o telarquia). Casi el 50% de las pubertades precoces se acompañan de adrenarquia prematura, pero la presencia de una adrenarquia prematura no implica necesariamente el inicio puberal.
ABORDAJE DIAGNÓSTICO DE LA ADRENARQUIA PREMATURA
Tradicionalmente, hablar de adrenarquia era sinónimo de pubarquia. Hoy día, aunque la pubarquia continua siendo la principal manifestación clínica, se considera que el signo precoz más sensible a la acción androgénica es el olor corporal apocrino. El término adrenarquia también implica cambios como cabello graso, seborrea cutánea, acné y comedones. Puede asociar aceleración de la velocidad de crecimiento con aumento de la maduración ósea, para lo que es esencial la valoración con una edad ósea (EO).
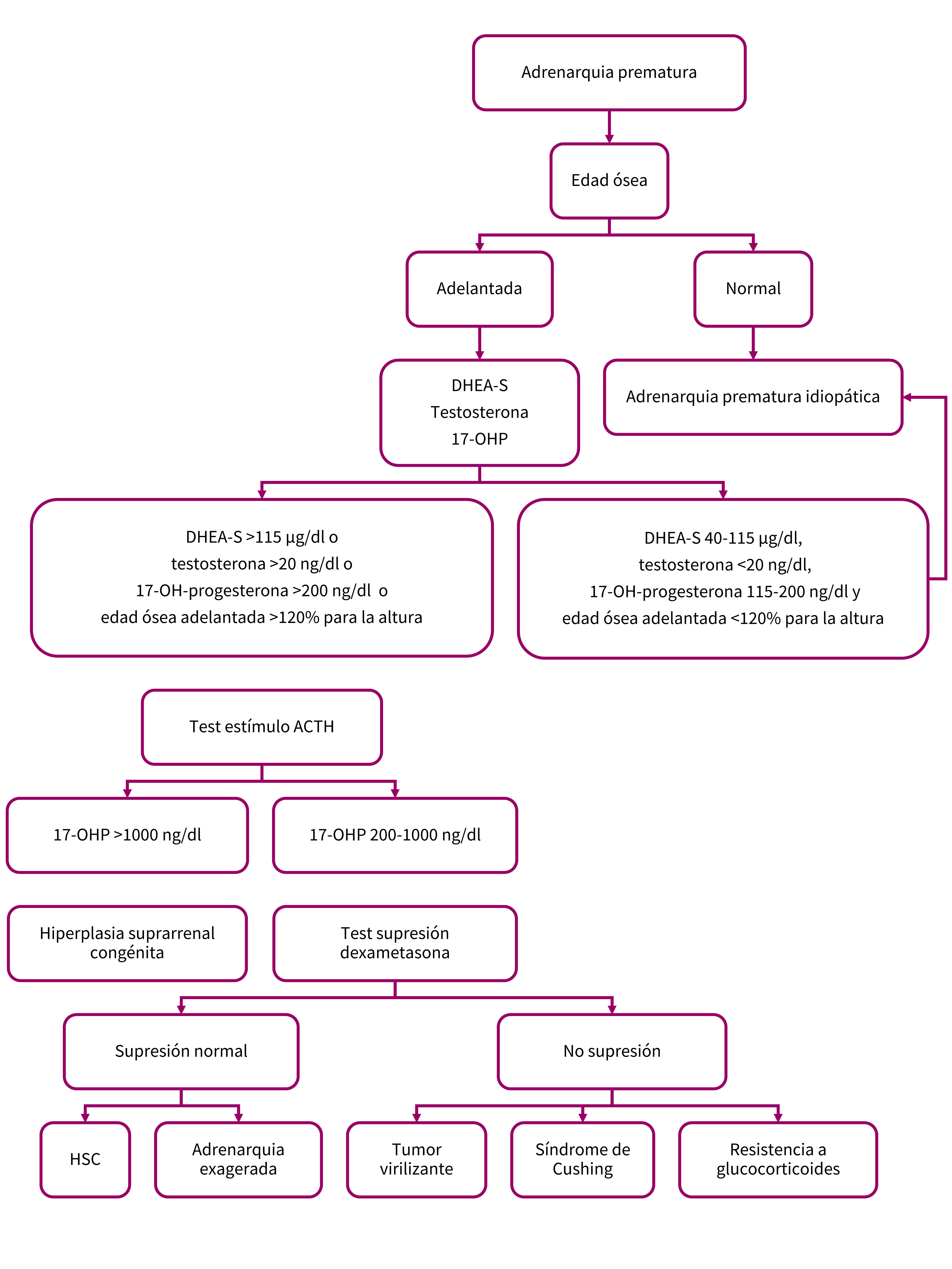
El diagnóstico requiere la demostración bioquímica de elevación de marcadores androgénicos séricos (DHEA-S >40 µg/dl) antes de los 8 años en niñas y de los 9 en niños (Figura 2).
Figura 2. Algoritmo diagnóstico de adrenarquia prematura. Mostrar/ocultar
Pruebas de primer nivel en la consulta de Pediatría
Anamnesis: la descripción de los signos y síntomas clínicos debe ser detallada (rapidez de instauración, tiempo de evolución, signos de virilización o pubertad), antecedentes personales (PEG, prematuridad, obesidad, enfermedades metabólicas…) y antecedentes familiares (etnia o raza, país de procedencia, datos del desarrollo puberal, enfermedades endocrinas, tratamientos…).
Exploración física completa: datos antropométricos con medición precisa de la talla, peso, índice de masa corporal y velocidad de crecimiento. Toma de presión arterial y descripción minuciosa del vello (distribución, características en color, grosor), del desarrollo mamario y genital (incluyendo palpación de testes).
Edad ósea: es la prueba diagnóstica básica más importante. Debe ser interpretada por un médico entrenado con experiencia.
- Si la EO es acorde a la edad cronológica (dentro de ±2 desviaciones estándar [DE] de la media), la predicción de la talla adulta (mediante métodos matemáticos como Bayley-Pinnaud) será normal. En tal caso, se tratará de una adrenarquia prematura idiopática con alta probabilidad, y no será necesaria la realización de otros test. Sin embargo, será importante reexaminar al paciente y repetir la edad ósea a los 6 meses para asegurar realmente que no es un trastorno virilizante de reciente aparición. Si la predicción de altura se mantiene estable, se pueden espaciar las peticiones de edad ósea en intervalos mayores.
- Si la EO es avanzada (>2 DE) es indicación de despistaje de trastornos virilizantes o alteraciones endocrinas. Es conveniente recordar que una edad ósea avanzada se asocia con más frecuencia a obesidad que a hiperandrogenismo. Los niños obesos suelen tener edad ósea significativamente adelantada para la edad cronológica pero siempre adecuada a la altura.
Pruebas de segundo nivel
En caso de existir signos importantes de virilización o si la EO está adelantada, es preciso determinar niveles basales de hormonas, por lo que convendría, llegados a este punto, derivar a consulta de Endocrinología Pediátrica.
Analítica básica: hemograma, bioquímica, perfil tiroideo, lipídico y del metabolismo hidrocarbonado. Si la adrenarquia prematura se acompaña de hipertensión arterial y sobrepeso, solicitar cortisol libre urinario en orina de 24 horas para despistaje de síndrome de Cushing.
Dehidroepiandrosterona-sulfato (DHEA-S): en la API los niveles de DHEA-S estarán por encima de los valores prepuberales, pero en rango de 40-115 µg/dl. Niveles de DHEA-S >700 µg/dl son característicos de la deficiencia de 3-β-hidroxiesteroide deshidrogenasa o de los tumores adrenales virilizantes (síntesis exclusivamente suprarrenal).
Testosterona: la API suele cursar con testosterona <20 ng/dl (0,7 nmol/l). Cifras elevadas sugieren patología tumoral gonadal pues la síntesis es prácticamente testicular u ovárica.
Androstendiona: síntesis suprarrenal y gonadal. En la API los valores son normales y ligeramente elevados en la HSC no clásica.
Hormona luteinizante (LH) y hormona foliculoestimulante (FSH): en niños con adrenarquia prematura parece prudente medir estas hormonas para descartar una verdadera pubertad precoz.
17-hidroxiprogesterona (17-OHP): de síntesis suprarrenal y ovárica. La determinación de 17-OHP basal es un buen test de cribado para la HSC. Realizar la extracción por la mañana temprano es muy importante, ya que los niveles de 17-OHP disminuyen rápidamente durante el curso del día. Los resultados se pueden interpretar de la manera siguiente:
- Niveles <115 ng/dl es lo normal en niños prepuberales y excluye la HSC no clásica con un 95% de confianza, aunque estos pacientes deberían ser vigilados clínicamente para comprobar que no aparecen otros signos de progreso puberal.
- Niveles de 17-OHP entre 115 y 200 ng/dl son acordes a una API.
- Niveles moderadamente elevados de 17-OHP (entre 200 y 1500 ng/dl) sugieren una posible HSC no clásica y es necesaria la confirmación diagnóstica con un test de ACTH.
- Niveles muy elevados de 17-OHP por encima de 1500 ng/dl son diagnósticos de HSC no clásica y no precisan confirmación mediante test de ACTH.
Pruebas de tercer nivel
La indicación de pruebas funcionales la establecerá el especialista en Endocrinología, no obstante, conviene conocerlas para la correcta interpretación de resultados.
Test de estimulación con ACTH: es la prueba definitiva para establecer el diagnóstico o excluir una HSC. Se solicita en pacientes con adrenarquia prematura y edad ósea adelantada si las pruebas complementarias arrojan resultados atípicos para una adrenarquia prematura idiopática (por ejemplo, DHEA-S >115 µg/dl, testosterona >20 ng/dl o elevación de 17-OHP matutina >200 ng/dl). El test se lleva a cabo usando ACTH 1-24 sintética (Cosyntropin®) intravenosa con dosis estándar de 250 µg y obteniendo muestras de sangre antes de la infusión de ACTH, 30 y 60 minutos después. Se determina la 17-OHP por su mayor rentabilidad diagnóstica, ya que la HSC por déficit de 21-hidroxilasa es la variante más frecuente. Se pueden medir también la 17-OH-pregnenolona y el 11-desoxicortisol si la sospecha es HSC por déficit de 11-β-hidroxilasa o 3-β-hidroxiesteroide deshidrogenasa. De rutina, además, se solicita la medición de cortisol para validar el test (cortisol >20 µg/dl). Niveles de 17-OHP >1000 ng/dl son sugestivos de déficit de 21-hidroxilasa y un pico >1500 es confirmatorio. Si el test de ACTH ha indicado una HSC, el tratamiento sustitutivo con glucocorticoides corregirá la patología y confirmará a su vez el diagnóstico.
Test supresión con dexametasona: se realiza administrando dexametasona a dosis de 1 mg/m2/día durante 4 días y en la mañana del quinto día se obtienen muestras de cortisol, DHEA-S y andrógenos. Sirve para distinguir entre un tumor virilizante (en el que no se consigue supresión androgénica pero el cortisol disminuirá) de otras formas de virilización como la HSC o la adrenarquia exagerada (donde existirá supresión del DHEA-S >75% y de cortisol). En el síndrome de Cushing o resistencia a glucocorticoides no se suprime ninguno normalmente.
Pruebas de imagen: la ecografía es el estudio radiológico inicial para despistaje de neoplasias adrenales, escrotales o pélvicas, aunque la capacidad de detección de neoplasias pequeñas es radiólogo dependiente. La tomografía computarizada y la resonancia magnética permiten una visualización más detallada de tumores.
Estudios genéticos: específicos según el déficit enzimático sospechado.
MANEJO DE LA ADRENARQUIA PREMATURA
El tratamiento de la adrenarquia prematura debe ser el mismo que el de la causa subyacente: extirpación quirúrgica de tumor virilizante, tratamiento médico sustitutivo con glucocorticoides en caso de HSC, retirada de fármaco en iatrogénicas, etc.
Si se trata de adrenarquia prematura idiopática no existe tratamiento específico, ya que es considerada como un proceso benigno variante de la normalidad.
Se debe informar y tranquilizar a los familiares, ya que estos niños tendrán una pubertad normal y alcanzarán una talla adulta acorde a la talla familiar. Conviene, además, explicarles los signos clínicos que indicarían virilización o pubertad precoz. Se proporcionarán consejos dietéticos y de ejercicio físico, pues la adrenarquia prematura se asocia a obesidad, síndrome metabólico y resistencia a insulina. Además, se podrá valorar el estudio de diabetes tipo 2 en los padres si hay historia familiar de esta patología.
Las niñas con adrenarquía prematura idiopática conllevan un riesgo aumentado del 15-20% de desarrollar SOP, por lo que se recomienda seguir a los pacientes hasta el final de la pubertad. Se puede aconsejar el uso de desodorantes o depilación y debemos estar muy pendientes a la repercusión psíquica que pueda ocasionar la adrenarquia en estos niños, ofreciendo soporte psicológico si lo precisan.
BIBLIOGRAFÍA RECOMENDADA
- Bezanilla C, Sastre A. Hiperandrogenismo. En: Guerrero Fernández J. Manual de diagnóstico y terapéutica en Endocrinología pediátrica. 1.ª ed. Madrid: Ergon; 2018. p. 666-77.
- Chrousos GP. Adrenal hyperandrogenism. En: UpToDate [en línea] [consultado el 01/03/2019]. Disponible en www.uptodate.com/contents/adrenal-hyperandrogenism#H21
- Novello L, Speiser PW. Premature adrenarche. Pediatr Ann. 2018;47:e7-e11.
- Rosenfield RL. Premature adrenarche. En: UpToDate [en línea] [consultado el 01/03/2019]. Disponible en: www.uptodate.com/contents/premature-adrenarche
- Utriainen P, Laakso S, Liimatta J, Jääskeläinen J, Voutilainen R. Premature adrenarche--a common condition with variable presentation. Horm Res Paediatr. 2015;83:221-31.
- Voutilainen R, Jääskeläinen J. Premature adrenarche: etiology, clinical findings, and consequences. J Steroid Biochem Mol Biol. 2015;145:226-36.
LECTURAS RECOMENDADAS
Para profundizar el tema de la hiperplasia suprarrenal congénita, os recomiendo este capítulo muy bien expuesto y de lectura rápida.
- Carcavilla A, Gracia Bouthelier R. Hiperplasia suprarrenal congénita. En: Guerrero Fernández J. Manual de diagnóstico y terapéutica en Endocrinología pediátrica. 1.ª ed. Madrid: Ergon; 2018. p. 763-78.
Es importante conocer los criterios diagnósticos de pubertad precoz tanto central como periférica. Aunque no es objeto de este tema, es recomendable su lectura para el diagnóstico diferencial con adrenarquia prematura.
- Pozo Román J, Muñoz Calvo MT. Pubertad precoz y retraso puberal. Pediatr Integral. 2015;XIX:389-410.