


¿Acné en un niño de 5 años? Hiperplasia suprarrenal no clásica
2 CS de Santa Bárbara. Toledo (España).
3 Complejo Hospitalario Universitario de Toledo. Toledo (España).
PUNTOS CLAVE
- El acné en niños prepuberales puede ser un signo clínico de adrenarquia que exige descartar la presencia de un hiperandrogenismo.
- La hiperplasia suprarrenal congénita no clásica, también llamada leve o tardía, agrupa un conjunto de trastornos hereditarios de la esteroidogénesis suprarrenal en las que el déficit enzimático es parcial.
- La principal diferencia respecto a la forma clásica es el momento en el que debuta la clínica y la gravedad de esta.
- Las manifestaciones clínicas suelen aparecer a partir de la tercera infancia y muchas veces pasan desapercibidas, sobre todo, en el caso de los varones.
- El diagnóstico definitivo se establece mediante estudio genético, el cual se realiza cuando existe un estudio hormonal sugestivo de la enfermedad.
- En la mayoría de los casos no se requiere tratamiento, aunque pueden ser útiles los corticoides u otros tratamientos según la sintomatología.
Este artículo enlaza con el artículo “Abordaje de la adrenarquia prematura en Atención Primaria”, publicado en el anterior número de FAPap, y desarrolla, ante un niño que presenta signos de adrenarquia, uno de los posibles diagnósticos diferenciales:
- La hiperplasia suprarrenal congénita no clásica (HSCNC).
- El síndrome de Cushing.
- Los tumores virilizantes.
- La adrenarquia precoz idiopática.
- El hiperandrogenismo suprarrenal funcional.
- Iatrogénica (exposición exógena a andrógenos).
CASO CLÍNICO
Niño de 5 años sin antecedentes de interés que consulta por fiebre de un día y medio de evolución asociada a odinofagia. En la exploración llama la atención la presencia de lesiones acnéicas, pápulas y comedones en la cara, con un estadio de Tanner prepuberal y sin olor apocrino del sudor corporal. Su peso era 23 kg (p85) y la talla 110 cm (p45).
No presentaba antecedentes personales de interés; las pruebas de cribado metabólico al nacimiento fueron negativas. La diversificación de la alimentación y desarrollo psicomotor habían sido correctos. Ante los datos de adrenarquia precoz, se solicita una radiografía de muñeca izquierda para determinar la edad ósea y posteriormente estudio analítico con hemograma, bioquímica y perfil hormonal con andrógenos para poder descartar un hiperandrogenismo.
INTRODUCCIÓN
La hiperplasia suprarrenal congénita (HSC) engloba un conjunto de trastornos de herencia autosómica recesiva de la esteroidogénesis suprarrenal del cortisol. En estas enfermedades se produce un bloqueo de alguna de estas rutas metabólicas debido al descenso de alguna actividad enzimática, lo que provoca una disminución de la producción de cortisol. Dicho descenso estimula la producción de corticotropina (ACTH) y esta, al aumentar su secreción, estimula la síntesis hormonal adrenal y la hiperplasia de la corteza adrenal que da nombre a la enfermedad.
La actividad enzimática puede verse afectada en distinto grado según la mutación que presente el paciente y este porcentaje de actividad enzimática determinará el grado de afectación clínica. Cuando la funcionalidad es inferior al 2%, aparecerá una forma clásica con afectación clínica desde el nacimiento; mientras que si la funcionalidad es superior al 30-50%, aparecerá una forma no clásica o tardía (HSCNC) con inicio de la clínica en la infancia, en la pubertad o incluso en la edad adulta o no dar manifestaciones clínicas (formas crípticas). Por tanto, la principal diferencia entre ambas formas es el grado de actividad enzimática residual.
PREVALENCIA
Mientras que las formas clásicas se estima que afectan a 1 de cada 14 000 personas (entre 1 de cada 280 o 28 000 según poblaciones), la HSCNC es mucho más prevalente. No existen datos muy precisos sobre la prevalencia real, debido en buena parte a que la clínica es poco expresiva en la mayoría de los pacientes. Se cree que podría afectar a 1 de cada 1000 individuos, siendo algo más prevalente en poblaciones de la cuenca del mediterráneo, los Balcanes o de ascendencia hispana o asquenazi (llegando hasta 1 de cada 100 individuos). A su vez, el estado de portador se estima que se puede presentar hasta en el 1% de la población (1 de cada 60-80 individuos), llegando al 10% en las poblaciones con mayor prevalencia.
CLÍNICA
La HSCNC podría clasificarse en formas sintomáticas y no sintomáticas. Estas segundas cursan únicamente con alteraciones hormonales, aunque puedan presentar signos clínicos leves de hiperandrogenismo. Las principales manifestaciones aparecen en:
Piel y alteraciones cutáneas
A nivel cutáneo, la piel grasa y el acné puede ser la primera manifestación clínica, como en el caso de nuestro paciente; aunque no suele ser el motivo de consulta más frecuente. Se debe sospechar en los casos de acné quístico refractario a tratamiento con retinoides y antibióticos, el cual está presente en un tercio de los pacientes pospuberales.
La pubarquia o axilarquia precoz, muchas veces asociada a olor apocrino del sudor, suele ser el motivo de consulta más frecuente. El hirsutismo o exceso de vello en zonas andrógeno-dependientes (cara, cuello, pecho, espalda, línea alba, ingles y muslos) aparece en hasta un 60% de las niñas, ascendiendo este porcentaje al 70% de las mujeres y 90% de las ancianas. Por el contrario, la alopecia podría afectar hasta un 8% de las pacientes afectas.
Pubertad y talla
El adelanto de la pubertad es casi una constante en la sintomatología de estos pacientes. La talla de los pacientes con HSCNC se encuentra influenciada, en gran parte, por el avance de la edad ósea asociado a un adelanto del inicio puberal. El cierre epifisario temprano puede determinar una talla final más baja. Sin embargo, aunque la media de la talla final se encuentra en torno al percentil 15 (-1 desviaciones estándar [DE]), esta no suele distar mucho de la talla genética salvo casos muy sintomáticos; incluso en ausencia de tratamiento, suele ser superior a la talla de las formas clásicas.
Aparato reproductor y fertilidad
Una de las diferencias respecto a las formas clásicas, es que no presentan ambigüedad ni virilización de genitales femeninos al nacimiento, aunque si puede aparecer clitoromegalia en un pequeño porcentaje de las pacientes.
Los problemas en la esfera reproductiva son más leves y menos frecuentes que en las formas clásicas. Se calcula que la HSCNC podría ser la causa de en torno al 4% de las mujeres que acuden a una consulta de fertilidad. Es más frecuente la subfertilidad que la infertilidad; pero también se ha asociado a un aumento de los abortos espontáneos.
En la adolescencia se puede presentar en forma de trastornos del ciclo menstrual en forma de amenorrea primaria (menos frecuente), amenorrea secundaria, oligomenorrea o ciclos anovulatorios. En algunas mujeres, la forma de presentación puede ser un síndrome de ovario poliquístico (SOP).
En los varones, las formas clásicas se han asociado al desarrollo de masas testiculares en forma de restos adrenales, que se asocian a oligoespermia e infertilidad. Sin embargo, aunque estos nódulos también pueden aparecer en algunos casos de HSCNC por norma general, las formas no clásicas tienen una función testicular normal y una fertilidad preservada.
ANAMNESIS Y EXPLORACIÓN FÍSICA
Respecto a los datos importantes que hay que recoger en la anamnesis de un niño con datos sugestivos de hiperandrogenismo prepuberal se encuentran la antropometría al nacimiento, las pruebas metabólicas neonatales, la ingesta de fármacos (valproico, fenitoína, ciclosporina, acetazolamida, glucocorticoides, penicilamina) y otras enfermedades endocrinas (obesidad, hiperprolactinemia, tumores gonadales y suprarrenales e insulinorresistencia). Un dato clave que nos debe de poner en alerta es si los síntomas han sido rápidamente progresivos, puesto que esto es sugestivo de tumores virilizantes. También es importante recabar datos sobre la familia, tanto de primer como de segundo grado; destacando en la madre la edad de presentación de la menarquia, la presencia de hirsutismo, irregularidades menstruales y problemas de fertilidad.
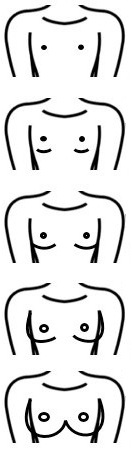
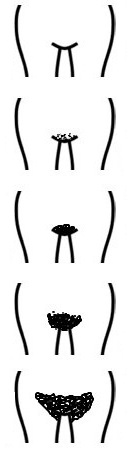
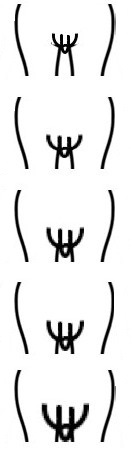
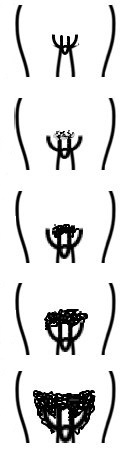
En la exploración física se deben recoger los datos de tensión arterial, antropometría (peso, talla e índice cintura-cadera), exploración general centrándose en la revisión de la piel, la palpación abdominal y la inspección de los genitales constatando si existe desarrollo puberal (estadios de Tanner) (Tabla 1).
Tabla 1. Estadios de Tanner. Mostrar/ocultar
PRUEBAS COMPLEMENTARIAS Y DIAGNÓSTICO DEFINITIVO
En base al diagnóstico diferencial, se debería solicitar una radiografía de mano y muñeca para determinar la edad ósea y un estudio hormonal que recoja hormonas tiroideas (TSH y T4L), cortisol libre urinario de 24 horas (si existe obesidad con talla baja o disminución de la velocidad de crecimiento), andrógenos (andostendiona, dihidroepiandrostendiona y testosterona total junto con la globulina fijadora de hormonas sexuales SGHB o la testosterona libre) y 17-hidroxiprogesterona (17-OHP).
La 17-OHP es un glucocorticoide que por medio de la acción de la enzima 21-hidroxilasa (21-0H, P450c21) se transforma en desoxicortisol (y posteriormente a cortisol mediante otras enzimas (Figura 1). El bloqueo en esta enzima por déficit funcional como sucede en más del 95% los casos de HSCNC, genera un acúmulo secundario del mencionado glucocorticoide.
Figura 1. Rutas metabólicas de la esteroidogénesis suprarrenal del cortisol simplificada. Mostrar/ocultar
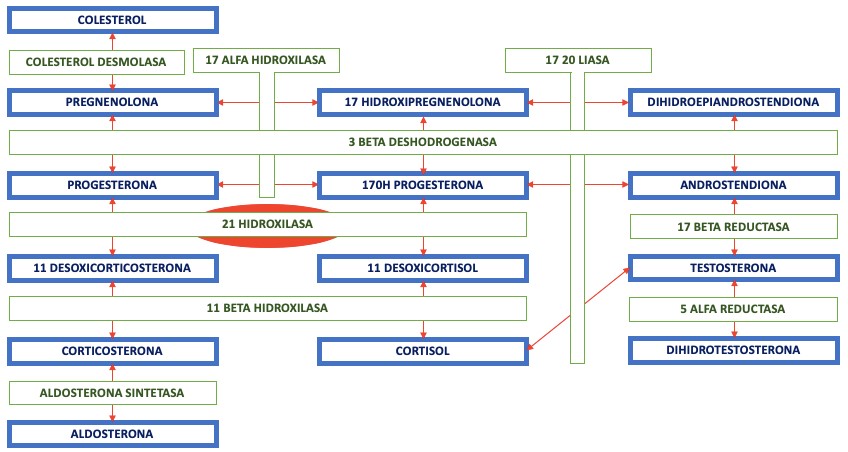
Los valores de la 17-OHP, a primera hora de la mañana, pueden orientar sobre la presencia de una HSCNC. Se debe de solicitar a todos los niños que presenten signos de adrenarquia precoz, edad ósea avanzada >2 años o aceleración de la velocidad de crecimiento para la edad. Valores de <1 ng/ml en pacientes prepuberales indicarían una adrenarquia precoz idiopática. En el caso de obtenerse valores >1 ng/ml en pacientes prepuberales y >2 ng/ml en puberales se debería realizar un test de estimulación con ACTH, siendo patológica si se produce una elevación >10 ng/ml. Las pruebas de imagen abdominal (ecografía, tomografía computarizada [TC], resonancia magnética [RM]) tienen su utilidad cuando se sospechan tumores virilizantes.
El diagnóstico definitivo se obtiene mediante el estudio molecular de las mutaciones conocidas que causan la HSCNC. La enzima más frecuentemente afectada es la 21-hidroxilasa codificada en el gen CYP21A2, región 6p21.33), de la que se han identificado 3 mutaciones situados en el exón 1 (Pro30Leu), 7 (Val281Leu) y 10 (Pro453Ser); debidas a la sustitución de nucleótidos (c.92C>T, c.844G>T y c.1360C>T respectivamente). Otra enzima mucho menos frecuentemente implicada es la 11-ß-hidroxilasa (P450c11ß, gen CYP11B1, región 8q24.3), que produce un aumento de los niveles plasmáticos de 11-desoxicortisol, 11-desoxicorticosterona e incluso 17-OHP. Esto genera que en ocasiones se diagnostica erróneamente de déficit de 21-OH, observando posteriormente que la genética no es compatible. Los estudios genéticos son útiles posteriormente para poder ofrecer el consejo genético a las familias. Los pacientes con formas no clásicas pueden ser a su vez ser portadores de mutaciones severas que en homocigosis o en heterocigosis compuestas dan lugar a formas clásicas.
El cribado neonatal de la HSC se realiza mediante una gota de sangre del talón del recién nacido a las 48-72 horas de vida. La elevación de la 17-OHP (generalmente >35 ng/ml) permite el diagnóstico de las formas clásicas, y aunque puede detectar algunas formas no clásicas, no es de utilidad para el cribado de las formas no clásicas.
TRATAMIENTO
El tratamiento se basa en el uso de corticoides a dosis más bajas que en la HSC clásica. El corticoide de elección, al igual que en las formas clásicas, es la hidrocortisona a dosis de 10-15 mg/m² repartidos en tres dosis. Aunque no hay indicaciones claras, se recomienda iniciar tratamiento en los casos que presenten aceleración de la velocidad de crecimiento con adelanto en la edad ósea de al menos 2 años o pacientes muy sintomáticos (acné grave, hirsutismo, irregularidades menstruales). Otros tratamientos se pueden usar en función de los síntomas o signos que vayan apareciendo (anticonceptivos orales, antiandrógenos, etc.). No se recomienda el tratamiento de niños asintomáticos.
Es importante que las familias comprendan la enfermedad y puedan tener a su alcance distintos recursos. Existe una asociación española de HSC (hiperplasiasuprarrenalcongenita.org) constituida a nivel nacional en 2013 que puede ser recomendada desde la consulta de pediatría de atención primaria. Su objetivo es aunar esfuerzos e iniciativas de familias y profesionales médicos en aras de mejorar la vida de pacientes afectados por este trastorno. Ofrece grupos de apoyo a las familias de nuestro país y proporciona información a las familias y enfermos.
RESOLUCIÓN DEL CASO CLÍNICO
En nuestro paciente se completó la anamnesis con los antecedentes familiares que hacían referencia al desarrollo puberal de los progenitores: la madre mide 161 cm (p49) y tuvo la menarquia a los 14 años, y su hermana y sobrina presentaron una pubarquia precoz. El padre, que mide 155 cm (p<1), refiere un estirón puberal adelantado.
Respecto a las pruebas complementarias solicitadas, la edad ósea presentaba un adelanto de un año.
En el estudio hormonal reveló:
- Elevación de los valores de la 17-OH-progesterona: 10,59 ng/ml (VN 4-7 años: 0,04-1,88).
-
Elevación de los valores de andrógenos: androstendiona 1,5 ng/ml (VN 4-7 años: 0,03-0,43).
DHEA: 218,6 µg/dl (VN 4-7 años: 2-46). - Las hormonas tiroideas fueron normales (TSH 4,409 uU/ml, T4 libre 1,16 ng/dl).
Debido al aumento de andrógenos se le realizó una ecografía abdominal, en la que no se observaron hallazgos relevantes; Con los resultados recibidos el diagnóstico de sospecha fue una HSCNC y fue derivado a la consulta de Endocrinología Pediátrica para la realización de un test de ACTH. Dicho test fue positivo, los valores de 17-OH-progesterona fueron de 16,37 ng/ml basal, 115,22 ng/ml a los 30 min. y 99,7 ng/ml a la hora de la estimulación. El diagnóstico definitivo se realizó posteriormente con el estudio molecular, mediante toma muestra genética de sangre periférica.
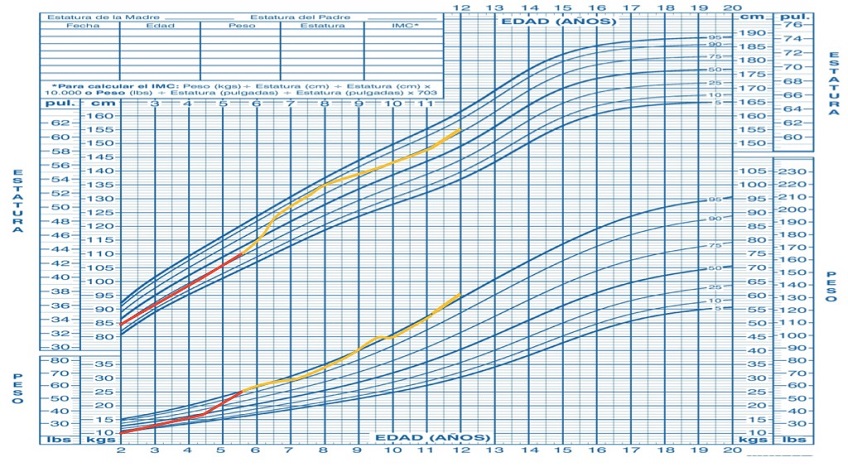
Nuestro paciente resulto ser homocigoto para la mutación Val281Leu; si bien en uno de los dos alelos, el heredado por parte materna, también presentaba otra mutación en heterocigosis asociada. Dichos datos fueron facilitados a la familia en la consulta de consejo genético. Nuestro paciente fue seguido de forma semestral y posteriormente anual en la consulta de Endocrinología Pediátrica. El peso se ha mantenido en percentil del 95; mientras que la talla subió hasta el percentil 90 con 8 años, descendiendo posteriormente por debajo del 75 desde los 9 años. El acné moderado ha sido la principal manifestación clínica de su patología. No ha necesitado recibir tratamiento con corticoides. Su talla diana familiar es de 164,5 cm. A los 12 años tiene un Tanner 4 y una talla de 155,7 cm (p71) (Figura 2).
Figura 2. Evolución de talla y peso de nuestro paciente. Mostrar/ocultar
Como en el caso de nuestro paciente, la HSCNC suele cursar como una enfermedad silente y no suele requerir tratamiento; sin embargo, su seguimiento es necesario en caso de que la evolución no sea favorable. La falta de expresión clínica o la no detección de la clínica sugerente hiperandrogenismo es el principal limitante para llegar al diagnóstico, que frecuentemente se realiza en la edad adulta.
BIBLIOGRAFÍA RECOMENDADA
- Alonso M, Ezequieta B. Hiperplasia suprarrenal congénita clásica o tardía. Rev Esp Endocrinol Pediatr. 2012;3:61-73.
- Asociación Española de Hiperplasia Suprarrenal Congénita. Disponible en: http://hiperplasiasuprarrenalcongenita.org
- Bezanilla López C, Sentchordi Montané L. Caracterización clínica, bioquímica y molecular de pacientes con hiperplasia suprarrenal congénita no clásica. Rev Esp Endocrinol Pediatr. 2015;6:7-11.
- Carcavilla A, Gracia Bouthelier R. Hiperplasia suprarrenal congénita. En: Guerrero Fernández J. Manual de diagnóstico y terapéutica en Endocrinología pediátrica. 1.ª edición. Madrid: Ergon; 2018. p. 763-78.
- Carmina E, Dewailly D, Escobar-Morreale HF, Kelestimur F, Moran C, Oberfield S, et al. Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum Reprod Update. 2017;23:580-99.
- Clemente León M, Escribano Muñoz A, Ezquieta Zubicaray B, Rodríguez Sánchez A. Hiperplasia suprarrenal congénita. Endocrinología Pediátrica. En: Continuum [en línea] [consultado el 17/06/2019]. Disponible en: https://continuum.aeped.es/modules/listado/647#.XQewHI9S_IU
- Guerrero López MC. Abordaje de la adrenarquia prematura en Atención Primaria. Form Act Pediatr Aten Prim. 2019;12;43-9.
- Labarta Aizpún JI, de Arriba Muñoz A, Ferrández Longás Á. Hiperplasia suprarrenal congénita. Protoc diagn ter pediatr. 2011;1:117-28
- Labarta JI, Bello E, Ruíz Echarri M, Rueda C, Martul P, Mayayo E, et al. Estado en la edad adulta y propuesta de optimización terapéutica de la hiperplasia suprarrenal congénita. An Pediatr (Barc). 2003;58:12-34.
- Nieman LK. Genetics and clinical presentation of nonclassic (late-onset) congenital adrenal hyperplasia due to 21-hydroxylase deficiency. En: UpToDate [en línea] [consultado el 17/06/2019]. Disponible en: https://www.uptodate.com/contents/genetics-and-clinical-presentation-of-nonclassic-late-onset-congenital-adrenal-hyperplasia-due-to-21-hydroxylase-deficiency
- Rodríguez Sánchez A, Sanz Fernández M, Echeverría Fernández M. Hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. Pediatr Integral. 2015; XIX:488-97.
- Sánchez Bachega T, Bilharino De Mendonca B. Hiperplasia suprarrenal congénita. En: Pombo M. Tratado de Endocrinología Pediátrica. 4.ª edición. Madrid: McGraw-Hill; 2010. p. 662-73.
- Sánchez Escudero V, García Escudero B, González Vergaz A, Bezanilla López C, Sentchordi Montané L, et al. Talla final en población pediátrica con la forma no clásica de hiperplasia suprarrenal congénita. Rev Esp Endocrinol Pediatr. 2017;8:20-8.
- Sancho Rodríguez ML, Bueno Lozano G, Labarta Aizpún JI, de Arriba Muñoz A. Evolución natural de la pubarquia precoz y posibles patologías asociadas. An Pediatr (Barc). 2018;89:238-45.
- Selma F Witchel, Azziz R. Nonclassic congenital adrenal hyperplasia. Inter J Pediatr Endocrinol. 2010;2010:625105.
- Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2018;103:4043-88.
- Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4133-60.
- Trapp CM, Oberfield SE. Recommendations for treatment of nonclassic congenital adrenal hyperplasia (NCCAH): an update. Steroids. 2012;77:342-6.