


Síndrome de Hunter o mucopolisacaridosis tipo II
2 MIR-Medicina Familiar y Comunitaria. Hospital Universitario Ramón y Cajal. CS Universitario Canillejas. Madrid. (España).
3 MIR-Medicina Familiar y Comunitaria. Hospital Universitario Ramón y Cajal. CS Universitario Canillejas. Madrid. (España).
PUNTOS CLAVES
- El síndrome de Hunter o mucopolisacaridosis tipo II es una enfermedad de almacenamiento lisosómico por déficit de la enzima iduronato-2-sulfatasa (I2S).
- Es una afección recesiva ligada al cromosoma X, afecta sobre todo a varones; una sola copia funcional en el gen X evita la enfermedad en mujeres.
- El déficit de la enzima I2S altera la descomposición de los mucopolisacáridos o glucosaminoglucanos (GAG), lo que lleva a la acumulación de dermatan sulfato (DS) y heparán sulfato (HS), en las células de todos los órganos.
- Los síntomas empiezan a ser evidentes a partir del primer año, la aparición de tosquedad facial es uno de los primeros signos.
- Ante un caso sospechoso, se determinarán GAG, HS y DS en orina simple en el paciente, teniendo en cuenta que hay falsos positivos y falsos negativos.
- Si el HS y el DS están elevados (o si son negativos y hay alta sospecha), se realizará biopsia de dermis para cuantificar la actividad de la I2S sérica de los leucocitos o fibroblastos de la dermis.
- El diagnóstico prenatal es posible y deseable en muestras de líquido amniótico o tejido de vellosidades coriónicas.
- En 2006 la FDA aprueba la idursulfasa (Elaprase®) como terapia de reemplazo enzimático de la enzima I2S, pero no atraviesa la barrera hematoencefálica.
- La terapia genética que reemplazase el cromosoma responsable sería la resolutiva, pero se encuentra aún en vías de investigación.
INTRODUCCIÓN
El síndrome de Hunter (SH) fue descrito por primera vez por el médico canadiense Charles Hunter en 1917. Pertenece al grupo de las mucopolisacaridosis (MPS) (síndromes de Hurler, Hunter, Scheie, San Filippo, Morquio, Sly), distinguiéndose estos síndromes entre sí por su mecanismo enzimático y genético. El SH es una mucopolisacaridosis tipo II (Tabla 1)1.
Tabla 1. Tipo de mucopolisacaridosis y otras variables1. Mostrar/ocultar
Es una enfermedad genética, recesiva ligada a X, determinada por una mutación en el cromosoma X (X*) en la región Xq28, que afecta a la función normal de la enzima iduronato 2 sulfatasa (I2S). El gen de la I2S es codificado por nueve axones y se han descrito más de una docena de mutaciones en este síndrome. Los pacientes con alteración importante del gen, y también los que presentan deleciones sufren una afectación grave, de tipo A; otros con mutaciones puntuales, sufren una afectación leve, tipo B. Afecta principalmente a varones y la mujer suele ser portadora. La homocigosis X*X* (ambos X afectos) es extraordinariamente rara, pues una sola copia funcional en el gen X evita el síndrome en mujeres; también hay descrito en literatura mundial algún caso de mujer Turner (45X*) y Hunter (donde no existe una copia sana de X funcional), e incluso se cita algún caso de mujer 46 X*X donde el X*es capaz de inactivar el alelo para la I2S activo del X2.
La I2S es esencial en la fragmentación y reciclaje de mucopolisacáridos específicos, conocidos también como glucosaminoglucanos (GAG), en este caso dermatán sulfato (DS) y heparán sulfato (HS). Al ser una enfermedad de almacenamiento lisosomal, los síntomas aparecen en todos los órganos progresivamente a lo largo de la vida, destacando de inicio la tosquedad facial que se observa a partir del año de vida.
La tasa de acumulación continuada varía según el grado de alteración funcional de la I2S, que puede ser parcial o totalmente inactiva: en algunos casos leves se han descrito personas sin retraso mental y con vida laboral activa. En las formas graves existe un retraso mental grave y expectativas de vida de 20 años o menos3.
PREVALENCIA
La incidencia es de 1 cada 130 000-170 000 nacimientos en Europa1.
CLÍNICA
La acumulación de DS y HS de manera continuada causa con el tiempo una disfunción orgánica multisistémica, variable en función del grado de alteración de la I2S. Si la afectación es leve, la sintomatología que a continuación se describe, se presentará de manera mucho más tardía y atenuada.
Crecimiento: puede que los “bebés Hunter” nazcan más grandes y crezcan más rápidamente en los primeros 12 o 15 meses. En la forma leve se puede alcanzar una talla de adulto normal en percentiles medios. Los afectados con más gravedad no suelen alcanzar una talla final mayor de 120-130 cm1,2.
El aspecto del recién nacido es normal. Es a partir del primer año cuando se ponen de manifiesto los primeros síntomas con cambios en sus rasgos faciales que se vuelven más toscos. Suelen tener la cara regordeta y la cabeza bastante grande con frente prominente. El cuello es corto y la nariz ancha con puente aplastado. Los labios suelen ser gruesos y la lengua grande, el esmalte dental muy frágil, el pelo abundante y espeso en cejas, cabeza y cuerpo (Figura 1).
Figura 1. Facies de síndrome de Hunter leve y grave7. Mostrar/ocultar

Piel: es gruesa, dura, con poca elasticidad. Alrededor de los omoplatos se pueden apreciar pequeños nódulos por acúmulo de GAG.
Otorrinolaringología: puente nasal aplastado, menor crecimiento de los huesos de la cara, aumento de adenoides y amígdalas, produciendo cuadros obstructivos y apneas respiratorias, favorecidos por un cuello corto y tráquea que se estenosa progresivamente y se ablanda por la textura anormal de los cartílagos, favoreciéndose las infecciones de repetición, faringoamigdalitis, rinitis, sinusitis y otitis.
También suele haber algún grado de sordera, de conducción, de transmisión o mixta. Si la trompa de Eustaquio queda bloqueada por el cúmulo de GAG, la mucosidad en el oído medio se acumulará y se volverá espesa como pegamento (“oreja de pegamento” o glue ear).
Oftalmología: degeneración retiniana, papiledema, glaucoma, pérdida de visión periférica, no suele haber opacidad corneal.
Tórax: rigidez que le dificulta realizar los movimientos respiratorios, estrechamiento de las vías respiratorias que, añadida al aumento de secreciones, provoca cuadros obstructivos respiratorios, con mayor riesgo de bronquitis y neumonía.
A nivel del corazón, las válvulas cardiacas se engrosan y estenosan, sobre todo la mitral y la aórtica, el miocardio aumenta en espesor y rigidez, pudiendo producir insuficiencia cardiaca congestiva, asimismo, las coronarias se estrechan, pudiendo haber episodios de ángor.
Abdomen: el hígado y el bazo se encuentran aumentados por almacenamiento de GAG, lo que provoca frecuentemente la aparición de hernias abdominales.
Alteraciones esqueléticas: como disóstosis múltiple, engrosamiento de huesos craneales, cierre prematuro de suturas, silla turca agrandada en forma de jota, clavículas cortas y engrosadas, espondilolistesis con o sin compresión medular, anomalías en huesos largos con acortamiento por engrosamiento de las metáfisis, hipoplasia de los núcleos de osificación, pelvis hipoplásica con cabezas femorales pequeñas, coxa valga, metacarpianos y metatarsianos acortados y ensanchados, con apariencia trapezoidal, síndrome del túnel carpiano, rigidez y contractura articular.
Sistema nervioso central: son muy frecuentes los síntomas derivados de la compresión medular en la región cervical, debido a hiperplasia de la duramadre y al engrosamiento del ligamento flavum. En la afectación grave, el almacenamiento progresivo de GAG en el cerebro produce retraso importante en el desarrollo intelectual a partir de los 2 años, regresión del desarrollo psicomotor con pérdida gradual de algunas habilidades, conservando otras. Si bien suelen ser alegres y cariñosos, a veces tienen un comportamiento terco o agresivo, siendo muy frecuente la hiperactividad. Puede haber hidrocefalia por bloqueo al paso del líquido cefalorraquídeo y cuadros convulsivos. Los afectados por la variante leve pueden tener una inteligencia normal1-4.
DIAGNÓSTICO
En el paciente con sospecha clínica de padecer SH, se debe evaluar una muestra de orina para determinar los niveles de GAG (Figura 2). Si el DS y el HS presentan cifras elevadas, se realiza la biopsia de dermis para analizar la actividad de la enzima I2S en leucocitos o fibroblastos, y si fuera deficitaria, ya con el diagnóstico de síndrome de Hunter, se realizaría estudio genético para conocer la mutación. Si los GAG fueran negativos, teniendo en cuenta la posibilidad de falsos negativos, se debería proseguir el estudio enzimático1,2,5.
Figura 2. Algoritmo diagnóstico en mucopolisacaridosis (MPS)1. Mostrar/ocultar
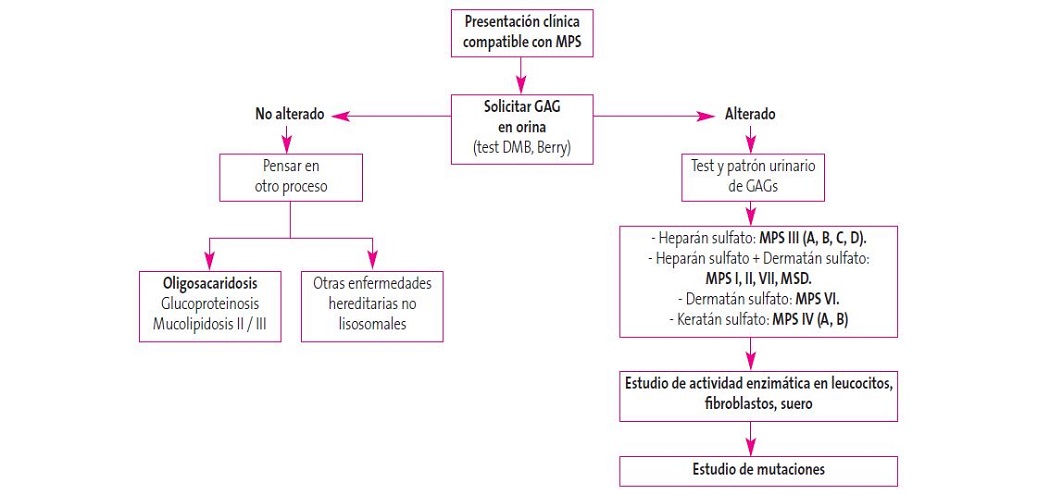
El análisis prenatal en familias afectadas permite que se pueda detectar si el feto varón lleva una copia defectuosa en estudio de microvellosidades coriónicas con biopsia de trofoblasto en la 11.ª a la 13.ª semanas de amenorrea y mediante amniocentesis en la semana 13.ª a 15.ª. En el feto femenino no es necesario realizar este estudio antes del nacimiento6.
TRATAMIENTO
Multidisciplinar: debe abordar las distintas complicaciones respiratorias (suelen necesitar amigdaloadenoidectomía), utilización de dispositivos de presión positiva continúa en vías respiratorias para resolver los cuadros obstructivos y las apneas del sueño. Además, precisan controles periódicos múltiples áreas especializadas (Cardiología, Traumatología, Rehabilitación, Ortopedia, Fisioterapia, Neurología, Psicología)1,2.
Terapia enzimática sustitutiva: la idursulfasa (Elaprase®), aprobada por la Food and Drug Administration (FDA) en el año 2006 en EE. UU., y en el 2007 por la Agencia Europea de Medicamentos (EMA), se administra una vez a la semana, por vía endovenosa, de por vida. Si se administra al diagnóstico y este es precoz, puede retrasar o incluso prevenir algunos de los síntomas del síndrome de Hunter. Es un medicamento de elevado coste, que no atraviesa la barrera hematoencefálica, por lo que no se resuelven los problemas neurológicos. Tampoco se han observado beneficios en las válvulas cardiacas ni a nivel óseo. Entre sus efectos secundarios se encuentran reacciones alérgicas, a veces graves1,2.
También se realiza el trasplante de células madre con células provenientes de médula ósea, como células de sangre de cordón umbilical. Precisa de una elevada histocompatibilidad HLA, no mejora sustancialmente los problemas musculoesqueléticos, aunque sí estabiliza los neurológicos, y mejora la afectación cardiaca y la patología ORL. La mortalidad está alrededor del 20%, dadas las complicaciones que del mismo puedan derivarse.
En fase II/III de experimentación en humanos se encuentra el tratamiento de sustitución enzimática vía intratecal, para intentar evitar la neurodegeneración.
La terapia génica sería la resolutiva, al reemplazar el cromosoma responsable de producir la enzima alterada, pero por ahora se encuentra en vías de investigación3,5,6.
CASO CLÍNICO
Acude a nuestra consulta de Pediatría una familia de refugiados sirios, compuesta por tres hijos varones de 5, 10 y 12 años y una hija de 13 años. Han llegado a Madrid procedentes de Turquía, donde se desplazaron 2 años antes desde Siria huyendo de la guerra civil. En Turquía, dos de sus hijos varones (a los 9 y 11 años) fueron diagnosticados de síndrome de Hunter, sin instaurar tratamiento, el tercer hijo varón está sano, y la hija es portadora del cromosoma afectado (Figura 3). Los dos hermanos presentan un síndrome de Hunter moderado (Figura 4). Están siendo atendidos desde su llegada en el Hospital Infantil Universitario Niño Jesús de Madrid, en el Servicio de Neurología, en la consulta de enfermedades neurodegenerativas y neurometabólicas y demás departamentos especializados.
Figura 3. Herencia ligada a X, en nuestro caso, de síndrome de Hunter. Mostrar/ocultar
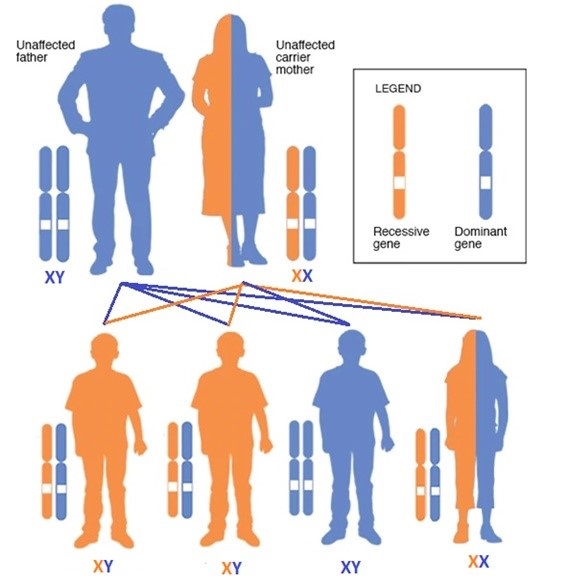
Figura 4. Facies y fenotipo de los pacientes de 10 y 12 años con síndrome de Hunter moderado. Mostrar/ocultar

Los dos hermanos afectos siguen tratamiento con Elaprase® semanal, por vía endovenosa, mediante port-a-cath, en dosis 0,5 mg/kg desde mayo de 2018.
En el control de las 50 semanas postratamiento, disminuyó el tamaño del bazo en un 90% en el hijo 1, normalizándose en el hijo 2; en ambos disminuyó el tamaño del hígado en un 80%, mejorando notablemente el volumen espiratorio forzado en 1 segundo y la capacidad vital forzada en ambos, sus valores urinarios de GAG se normalizaron hasta quedar por debajo de los límites superiores de los valores normales. En los dos hermanos disminuyó la hipertrofia ventricular izquierda que presentaban al inicio del tratamiento, pero no ha habido mejoría en la afectación de las válvulas cardiacas, siendo la mitral la más estenosada. Ha disminuido notablemente la contractura y rigidez articular. Han sido adenoideamigdalectomizados en enero 2019, y el hijo 2 fue intervenido en mayo 2019 del túnel carpiano. Siguen tratamiento también con Enalapril® para controlar su hipertensión arterial.
La hermana portadora deberá recibir consejo genético cuando se encuentre en edad de reproducción.
EVOLUCIÓN
Se han logrado avances importantes en los últimos años con la terapia enzimática sustitutiva, se ha aumentado la supervivencia tras el trasplante de médula, pero se debe insistir en el diagnóstico precoz y la investigación en genética preimplantatoria, con fármacos capaces de saltarse los codones de parada prematura o que impidan que la enzima alterada se degrade, con la esperanza de que la terapia genética se haga realidad en un futuro no muy lejano1.
DIRECCIONES DE INTERÉS
- Asociación MPS España (www.mpsesp.org/portal1/a_index.asp). Entidad declarada de utilidad pública.
- Consulta de enfermedades neurodegenerativas y neurometabólicas. Servicio Neurología. Hospital Infantil Universitario Niño Jesús. Dr. Luis González Gutiérrez-Solana. Teléfono: 91 503 59 00.
BIBLIOGRAFÍA
- González-Meneses López A, Barcia Ramírez A, Díaz Rodríguez J. L. Protocolo de actuación en la mucopolisacaridosis. Protoc diagn ter pediatr. 2010;1:24-36.
- Burton BK, Giugliani R. Diagnosing Hunter syndrome in pediatric practice: practical considerations and common pitfalls. Eur J Pediatr. 2012;171:631-9.
- Guillén-Navarro E, Javier Blasco A, Gutiérrez-Solana LG, Luz Couce M, Cancho-Candela R, Lázaro P. Guía de práctica clínica para el tratamiento del síndrome de Hunter. Med Clin (Barc). 2013:141:453.el-453.e13.
- Arias Eulate JC, Angulo Flores MD, Rueda Muñoz Z, Paz G. Síndrome de Hunter mucopolisacaridosis (II): reporte de un caso. Rev Cient Cienc Méd. 2011;14:40-2.
- Tejeda Dilou Y, del Río Monier Y, Álvarez Valiente H, Coca Prádez D, Núñez Copo AC. Síndrome de Hunter. Asesoramiento a parejas y familiares con riesgo. MEDISAN. 2013;17:4045-50.
- Scarpa M. Mucopolysaccharidosis Type II. En: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. (eds.). GeneReviews® [en línea] [consultado el 12/03/2020]. Disponible en: www.ncbi.nlm.nih.gov/pubmed/20301451
- MPS II o Síndrome de Hunter. Guía práctica para entender la enfermedad. En: Asociación MPS España [en línea] [consultado el 12/03/2020]. Disponible en www.mpsesp.org/portal1/images/content/Guia%20Hunter.mail.pdf