

Displasia espondiloepifisaria tardía: importancia de la talla baja en el niño mayor
2 MIR-Cirugía Ortopédica y Traumatología. Complejo Hospitalario Universitario de Albacete. Albacete. (España).
3 Servicio de Neuropediatría. Complejo Hospitalario Universitario de Albacete. Albacete. (España).
PUNTOS CLAVE:
- En las revisiones del Programa de Salud Infantil es importante la valoración auxológica, incluyendo las proporciones corporales para diferenciar el hipocrecimiento en armónico o disarmónico.
- No todas las osteocondrodisplasias son desproporcionadas. Sin embargo, una talla baja desproporcionada siempre es patológica.
- Existen displasias óseas de aparición tardía, las cuales se detectan por un descenso en la velocidad de crecimiento y en la talla a partir de los 6 años de edad.
- La displasia espondiloepifisaria tardía es una entidad que afecta a varones y de forma predominante a la columna vertebral, producida por una mutación del gen TRAPPC2.
- Se caracteriza por talla baja desproporcionada de aparición a los 6-8 años en un niño con una talla previa normal junto con anomalías óseas como escoliosis, tórax en barril y platispondilia (reducción de la altura de una o varias vértebras).
- Es importante pensar en esta patología cuando tengamos un descenso importante en el percentil de talla o velocidad de crecimiento en el niño mayor asociado a las alteraciones comentadas.
- Su tratamiento es ortopédico o quirúrgico. Se basa en corregir deformidades y prevenir lesiones musculoesqueléticas. Sin embargo, no existe un tratamiento curativo.
INTRODUCCIÓN
Hablamos de talla baja o hipocrecimiento cuando se cumple alguno de los siguientes criterios: talla por debajo de -2 desviaciones estándar (DE), una velocidad de crecimiento (VC) inferior a -1 DE para la edad y sexo mantenida más de 2-3 años, talla por debajo de 2 DE del carril de crecimiento de su talla diana, o expectativas de talla adulta más de 2 DE por debajo de la talla diana1. Los hipocrecimientos pueden ser de causa desconocida (80% de los casos; dentro de los cuales se incluyen las variantes normales de talla baja) o de causa conocida (20% de ellos; como osteocondrodisplasias, retraso de crecimiento intrauterino, cromosomopatías, malnutrición, etc.).
¿Cuándo sospechar una talla baja patológica?
Para realizar un diagnóstico de talla baja es necesaria como orientación inicial una valoración auxológica completa, incluyendo las proporciones corporales (segmento superior, segmento inferior, envergadura y las relaciones entre ellas). Esta primera evaluación ayuda a diferenciar si una talla baja es armónica o disarmónica (es decir, con desproporción de los segmentos corporales), siendo esta última siempre patológica y característica de las osteocondrodisplasias. Además, se deben analizar las curvas pondero-estaturales desde el nacimiento y comparar la edad ósea con la edad cronológica (EC).
Una vez orientada la línea diagnóstica y según nuestra sospecha clínica, debemos completar el estudio con pruebas complementarias de primer nivel: hemograma, bioquímica con perfil lipídico, función hepática, renal y tiroidea, anticuerpos antitransglutaminasa; y de segundo nivel: perfil hormonal con IGF-I, IGFBP-3, insulina basal, hormona luteinizante (LH), hormona folículoestimulante (FSH); estudio radiológico completo; cariotipo o genética, entre otros.
OSTEOCONDRODISPLASIAS
Las osteocondrodisplasias suelen ser de causa genética y son enfermedades que cursan con malformaciones en la estructura, densidad, tamaño o forma de huesos o cartílagos. Tienen una incidencia de 15,7/100 000 recién nacidos2. Su amplia clasificación varía rápidamente gracias a los avances genéticos, existiendo más de 450 entidades descritas1. Depende de la zona topográfica que afecten, distinguimos displasias epifisarias, fisarias, metafisarias o diafisarias.
Su máxima expresión clínica se da en la infancia y adolescencia, sin embargo, en los casos graves ya se aprecia desde el nacimiento. En cuanto al tratamiento, se basa en el ortopédico y el quirúrgico para corregir deformidades, prevenir lesiones (esqueléticas o neurológicas) y en algunos casos, alargamiento de extremidades. No existe tratamiento curativo, aunque están en investigación algunas terapias (como la sustitución enzimática en enfermedades de depósito)3.
CASO CLÍNICO
Varón de 12 años con talla baja. No presenta antecedentes personales de interés: parto eutócico a las 40 semanas de edad gestacional, peso al nacimiento de 3200 gramos (P31, -0,5 DE), longitud de 50 cm (P41, -0,23 DE), con proporciones corporales normales, perímetro craneal de 34 cm (P25, -0,68 DE); periodo perinatal y desarrollo psicomotor normales.
En las visitas que realiza dentro del Programa de Salud Infantil en su centro de salud se aprecian unas curvas de crecimiento pondero-estatural y de velocidad de crecimiento normales hasta los 8 años, presentando en los últimos 4 años una caída importante de percentil de talla, desde el P50 al P3-10. En la exploración física presenta: peso 37,5 kg (P14, -1,11 DE), talla: 138,2 cm (P<1, -2,37 DE), brazada 144 cm y VC de 2,6 cm/año (P<1, -3,43 DE). Destaca talla baja a expensas de un segmento superior acortado, con tórax en tonel, cuello corto, test de Adams patológico e hiperlordosis lumbar. Desarrollo puberal acorde a su edad.
Se realiza estudio de talla baja: bioquímica con perfil hepático, renal y tiroideo normal; anticuerpos antitransglutaminasa negativos; perfil hormonal (IGF1: 213 ng/ml, IGFP3: 4,6 ng/ml, insulina basal: 3 mcU/ml, LH: 2,7 mUI/ml, FSH: 2,34 mUI/ml) con valores normales para edad y sexo; edad ósea de 13 años (que se corresponde con la EC). A la vista de los resultado analíticos normales y de las características de la exploración física, se solicita un estudio radiográfico completo donde se aprecia platispondilia, hiperlordosis lumbar, escoliosis leve (ángulo de Cobb menor de 20°) y dismetría de miembros inferiores (Figuras 1 y 2). Ante la asociación de talla baja desproporcionada de manifestación tardía y las anomalías esqueléticas descritas, se sospecha displasia espondiloepifisaria tardía y se solicita estudio genético, obteniéndose la variante patogénica c.93+5G>A en el gen TRAPPC2 en homocigosis, ligada al cromosoma X. Se realiza estudio genético familiar, siendo la madre portadora de la misma mutación.
Figura 1. Radiografía anteroposterior de pelvis: ensanchamiento de la cabeza femoral bilateral y dismetría de miembros inferiores (derecho acortado). Mostrar/ocultar
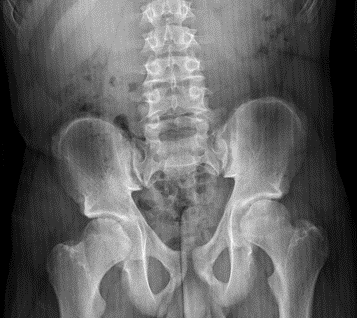
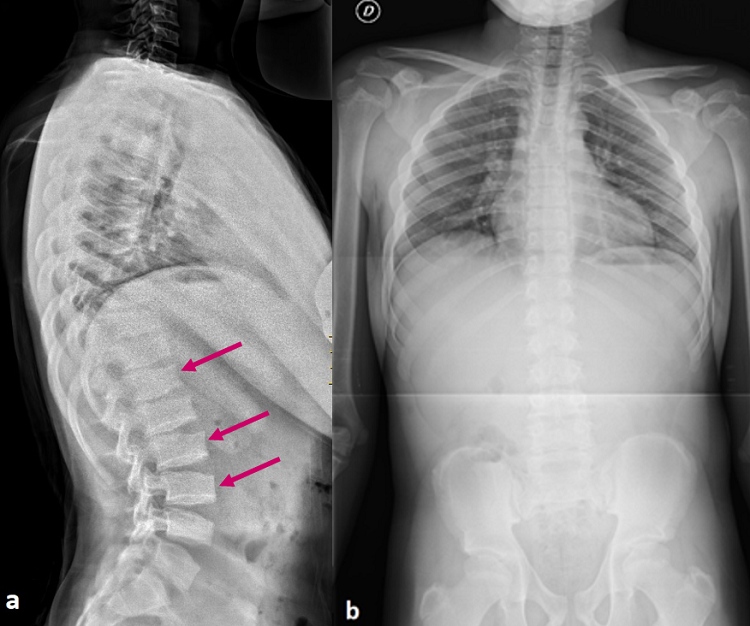
Figura 2. a) Radiografía lateral de columna vertebral: se observa platispondilia (cuerpos vertebrales planos) con ¨jorobas¨ en los platillos inferiores y superiores de los cuerpos vertebrales (flechas) e hiperlordosis lumbar. b) Radiografía anteroposterior de columna vertebral: escoliosis leve y asimetría de hombros compensatoria. Mostrar/ocultar
Actualmente el paciente tiene 16 años y su somatometría es: peso 47 kg (P7, -1,52 DE), talla: 148,7 cm (P<1, -3.38 DE) y brazada de 160 cm.
DISPLASIA ESPONDILOEPIFISARIA TARDÍA
Las osteocondrodisplasias pueden ser congénitas, las más frecuentes y graves, con manifestaciones clínicas desde el nacimiento e incluso prenatales; o de aparición tardía, las cuales se presentan en el niño mayor y adolescente, sin expresión clínica al nacimiento y primeros años de vida. Una de ellas es la displasia espondiloepifisaria tardía.
¿En qué consiste?
Se produce por una mutación en el gen TRAPPC2 (anteriormente conocido como SEDL)4. Presenta una herencia ligada a X, por lo que la enfermedad va a afectar a los varones, mientras que las mujeres serán portadoras. En un 20% de los casos no se encuentra esta mutación.
¿Cuáles son sus manifestaciones clínicas? ¿Cómo se diagnostica?
Se caracteriza por una afectación predominante de la columna vertebral, por lo que produce una talla baja desproporcionada por un tronco corto en tonel, siendo típica una brazada 10-20 cm superior a la altura que se detecta hacia los 6-8 años, con una talla final de 137-163 cm, presentando previamente una curva y velocidad de crecimiento, así como una longitud y proporciones corporales al nacimiento normales4. Entre otras manifestaciones clínicas, destacan la hiperlordosis lumbar, cuello corto, escoliosis, afectación de grandes articulaciones y nódulos en los dedos por tumefacción de las articulaciones interfalángicas, siendo el desarrollo motor y cognitivo normales. Estas anomalías producen dolor lumbar y articular, siendo típica la artrosis precoz de cadera. En la radiografía ósea son típicas la platispondilia con jorobas superiores e inferiores y alteraciones de las caderas similares a la enfermedad de Perthes4.
El diagnóstico de confirmación se realiza mediante estudio genético dirigido, pero antes es preciso sospecharlo al enfrentarse a los hallazgos clínicos y radiológicos, es decir, pensar en esta patología ante un niño mayor o adolescente con descenso en la velocidad de crecimiento y talla baja desproporcionada, con un tronco corto junto con las anomalías esqueléticas descritas, que hasta esa edad presentaban una curva de talla normal.
Es importante conocer que existen displasias óseas de aparición tardía. Muchas de ellas, además de la afectación en la talla y del desarrollo esquelético pueden asociarse con patologías a otros niveles, como cardiaco u oftalmológico. Un diagnóstico correcto permitirá realizar un seguimiento completo de todas las alteraciones. Además, el diagnóstico es imprescindible para realizar un buen consejo genético tanto de los portadores como de los pacientes afectos.
BIBLIOGRAFÍA
- Pozo Román J. Crecimiento normal y talla baja. Pediatr Integral. 2015;XIX:411.e1-411.e23.
- Bacino C. Skeletal dysplasias: Specific disorders. En: UpToDate [en línea] [consultado el 17/12/2020].Disponible en: www.uptodate.com/contents/skeletal-dysplasias-specific-disorders
- Delgado A. Manual Curso COT. 5.ª edición. Madrid: Panamericana; 2017-2018.
- Tiller GE. X-Linked Spondyloepiphyseal Dysplasia Tarda. En: GeneReviews® [en línea] [consultado el 17/12/2020]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1145/