

Diagnóstico y tratamiento de la enfermedad de Kawasaki en la infancia
2 Pediatra. EAP Fraga. Fraga. Huesca (España).
3 FEA de Pediatría. Unidad de Enfermedades Infecciosas. Servicio de Pediatría. Hospital Universitario Miguel Servet. Zaragoza. (España).
PUNTOS CLAVE
- La enfermedad de Kawasaki (EK) es una entidad poco frecuente que supone la principal causa de enfermedad coronaria adquirida en la infancia en los países desarrollados.
- Su diagnóstico se basa en la reunión de datos clínicos o analíticos, carece de un signo o síntoma clínico patognomónico, ni una prueba que lo diagnostique, por lo que precisa una alta sospecha clínica.
- Debe plantearse en el diagnóstico diferencial de cuadros más frecuentes, y se debe plantear en todo cuadro febril de ≥5 días evolución sin una causa que la justifique.
- Su diagnóstico es poco probable si tras 7 días de fiebre no se produce elevación de los reactantes de fase aguda (proteína C reactiva [PCR], procalcitonina [PCT] o velocidad de sedimentación globular [VSG]) ni trombocitosis.
- Existen formas completas, incompletas y atípicas, por lo que ante su sospecha y en ausencia de justificación para cuadro febril, se debe plantear el tratamiento para disminuir el riesgo de afectación coronaria.
- Es fundamental la valoración y seguimiento cardiológico.
- Ante su sospecha, se debe pautar tratamiento en los primeros 10 días de la enfermedad para disminuir el riesgo de afectación cardiaca. También debe tratarse si se diagnostica más tarde, si persiste la inflamación sistémica, manifestada por la elevación de reactantes de fase aguda (PCR o VSG), junto con persistencia de fiebre no atribuible a otra causa o alteraciones coronarias.
- La primera línea de tratamiento se basa en la administración de inmunoglobulinas (Ig) y ácido acetilsalicílico (AAS) a dosis antiinflamatorias. Pudiéndose asociar corticoides en las formas graves y en las de alto riesgo de resistencia a las Ig.
- En la reciente pandemia de SARS-CoV-2 se ha descrito en niños un cuadro inflamatorio sistémico con rasgos similares a Kawasaki asociando clínica digestiva, shock y mayor elevación de los reactantes de fase aguda.
RESUMEN
La enfermedad de Kawasaki es una vasculitis sistémica autolimitada. Su etiología continúa siendo desconocida. Supone la causa más frecuente de afectación coronaria cardiaca adquirida en la infancia (con riesgo de producir aneurismas coronarios, infarto agudo de miocardio y muerte súbita). Su diagnóstico se basa en la reunión de criterios clínicos. Una cuarta parte de las formas pueden ser incompletas. Su tratamiento consiste en la administración de inmunoglobulinas endovenosas con antiagregantes, que administrados en los 10 primeros días de evolución de la enfermedad disminuyen el riesgo de afectación cardiaca a menos del 5%. Las formas refractarias precisan la administración de corticoides y/o otros tratamientos biológicos.
Durante la pandemia de la COVID19 se han descrito casos pediátricos con criterios clínicos similares a la enfermedad de Kawasaki con/sin shock, y al síndrome del shock tóxico en pacientes con infección pasada por SARS-CoV-2.
INTRODUCCIÓN
La enfermedad de Kawasaki, conocida previamente como síndrome linfomucocutáneo fue descrita en Japón por Tomisaku Kawasaki en 1967. Es una vasculitis aguda autolimitada de vasos de mediano y pequeño calibre. Supone la causa más frecuente de enfermedad cardiaca adquirida en la infancia en los países desarrollados y la segunda causa de vasculitis en la infancia tras la púrpura de Schönlein-Henoch1,2. Hasta un 25% de los pacientes no tratados con inmunoglobulinas intravenosas (IgIV) desarrollan alteraciones coronarias, que desciende al 4% de los tratados2.
Es más frecuente en los países asiáticos, alcanzando en Japón una incidencia de 265 por 100 000 menores de 5 años, en Estados Unidos se sitúa en torno a 25 por cada 100 000 menores de 5 años3 y en Europa en torno 5,4-15 por cada 100 000 menores de 5 años.
En el 85% de los casos ocurre en menores de 5 años, con una máxima incidencia entre los 18 y 24 meses. Es menos frecuente en menores de 3 meses y en mayores de 5 años, teniendo estos grupos mayor riesgo de desarrollar alteraciones coronarias. Ocurre con más frecuencia en varones 1,5:12.
ETIOLOGÍA
La causa continúa siendo desconocida hoy en día. Se trata de una respuesta inmune que se cree que puede estar iniciada por uno o varios desencadenantes transmisibles (apoyado por el incremento estacional de casos, porque se presenta en ocasiones de forma epidémica y la asociación de fiebre, exantema, adenopatía y mucositis como en otros cuadros infecciosos). Se produciría una respuesta autoinmune contra la pared de los vasos sanguíneos en personas genéticamente predispuestas. Su baja frecuencia en los primeros meses de la vida y en la edad adulta, sugeriría que estos últimos estarían inmunizados, y la posibilidad de protección en primeros meses de la vida por trasferencia de anticuerpos maternos vía placentaria2.
MANIFESTACIONES CLÍNICAS4
Se describen tres fases clínicas: aguda, subaguda y convalecencia.
Fase aguda (primeros 10 días):
- Fiebre: aparece en el 100% de los casos, su inicio determina el primer día de enfermedad. Suele ser elevada (39-40 °C).
- Afectación de extremidades: edema doloroso en dorso de manos y pies y eritema bilateral palmo plantar. Se produce descamación periungueal en fase subaguda. En 1-2 meses pueden aparecer líneas transversales en uñas (líneas de Beau).
- Afectación ocular: aparece entre el 2-4.º día de fiebre en el 90-95% de los casos. Consiste en hiperemia conjuntival sin exudado, con un halo circundante más claro alrededor el iris.
- Exantema: polimorfo, confluyente sin vesículas, ni costras, ni petequias. Con más frecuencia es maculopapular inespecífico (90-92%), aunque puede ser purpúrico, urticarial, escarlatiniforme, eritrodérmico o similar al eritema multiforme. Localizado en tronco, abdomen y extremidades. Puede ser más intenso en región perineal, con descamación temprana.
- Afectación de labios y mucosas: eritema, sequedad, descamación, fisuras, grietas o sangrado espontáneo (92%) en labios. Lengua aframbuesada con papilas prominentes. Puede asociar eritema orofaríngeo sin exudados ni ulceraciones.
- Adenopatía (50-52%): suele ser unilateral, firme, fluctuante, de localización cervical anterior y clásicamente mayor de 1,5 cm.
-
Otras manifestaciones clínicas menos frecuentes pueden ser a nivel:
- Genitourinario: uretritis con piuria estéril (hasta 60%).
- Articular: en la primera semana (30%).
- Digestivas: dolor abdominal, diarrea, vómitos y dilatación (hidrops) vesicular (15%)
- Sistema nervioso: irritabilidad y somnolencia, meningitis aséptica (25%) e hipoacusia neurosensorial.
- Otras: afectación testicular, efusión pleural, nódulos pulmonares, síndrome hemofagocítico.
- Fase subaguda (días del 11 al 25): desaparición de signos clínicos previos y aparición de trombocitosis y descamación periungeal a las 2-3 semanas de inicio de la fiebre.
- Convalecencia (hasta el día 60): hasta normalización de reactantes de fase aguda y trombocitosis.
CRITERIOS DIAGNÓSTICOS
No existen manifestaciones clínicas o pruebas diagnósticas patognomónicas, se diagnostica por criterios clínicos, apoyando su diagnóstico los marcadores inflamatorios de laboratorio. No todos los criterios tienen porque estar presentes a la vez. En caso de no cumplir criterios de EK completa, pero tener anomalías coronarias, se confirma el diagnóstico. La normalidad cardiaca en la primera semana no excluye el diagnóstico.
-
EK completa: fiebre ≥5 días y ≥4 criterios clínicos simultáneos o sucesivos (exantema, afectación de extremidades, afectación ocular, mucositis y adenopatía) (Tabla 1).
Tabla 1. Diagnóstico de enfermedad de Kawasaki típico o completo. Mostrar/ocultar
-
EK incompleta: fiebre ≥5 días de causa inexplicable y 2 de los 5 criterios clínicos. La afectación cardiaca puede ser confirmatoria (Figura 1).
Figura 1. Sospecha de Kawasaki incompleto. Mostrar/ocultar
- EK atípica se reserva para cuando hay manifestaciones atípicas (nefritis, shock cardiogénico, síndrome hemofagocítico secundario), independientemente sea completo o no.
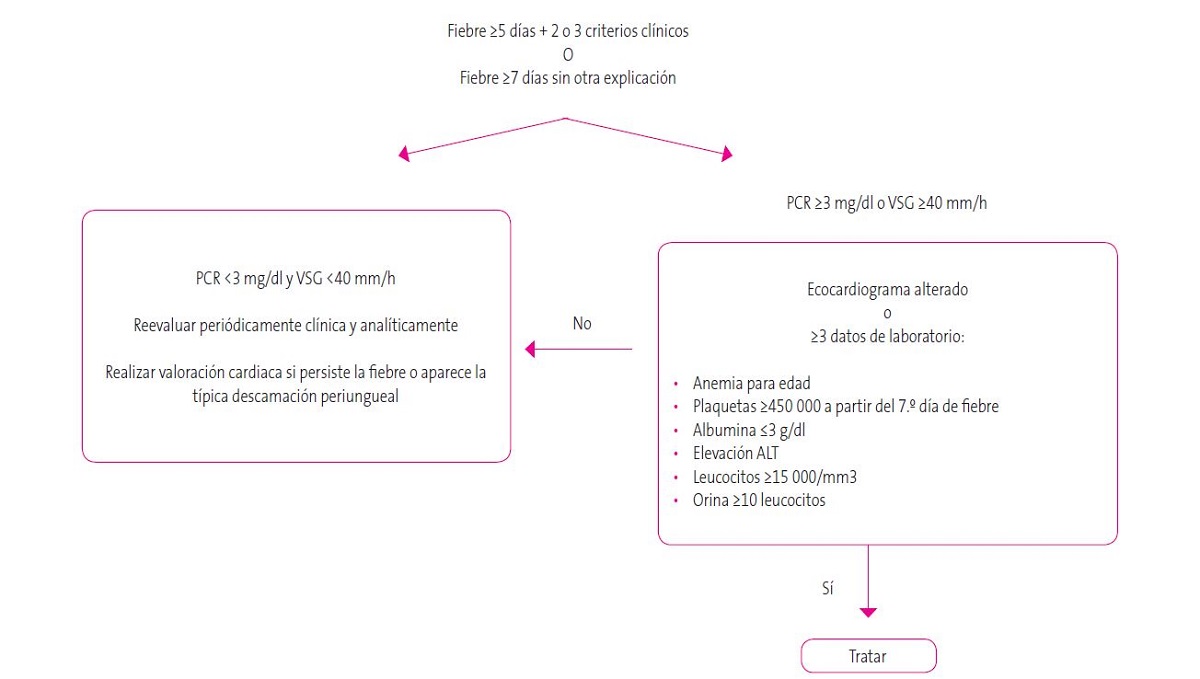
En caso de presentar fiebre y menos de 4 criterios clínicos principales, con datos de laboratorio o cardiológicos compatibles, considerar enfermedad incompleta o atípica.
Datos de laboratorio que sirven para apoyar el diagnóstico:
- Leucocitosis (≥15 000/mm3) con neutrofilia (≥10 000/mm3).
- Elevación reactantes de fase aguda PCR (≥30 mg/l), PCT o VSG (≥40 mm/h), tras administración de las IgIV la elevación de la VSG no se debe usar como seguimiento del proceso inflamatorio, pues puede verse faltamente elevada5.
- Anemia normocítica y normocrómica durante el proceso agudo y que se resuelve espontáneamente. Trombocitosis (≥450 000) reactiva a partir del 7 día que en casos no complicados se normaliza en 4-8 semanas, aunque en ocasiones en la primeras 2 semanas puede haber trombopenia6.
- Elevación triglicéridos y disminución colesterol.
- Hiponatremia.
- Elevación transaminasas, GGT y bilirrubina en 40-67% de los pacientes.
- Hipoalbuminemia (≤3 g/dl).
- Líquido cefalorraquídeo (LCR): meningitis aséptica, predominio de mononucleares, con glucosa y proteínas normales.
- Troponinas y pro-BNP. Indicadores de daño miocárdico.
Resulta fundamental el estudio cardiológico (electrocardiografía [ECG] y ecocardiografía): que debe realizarse en el momento del diagnóstico, pero nunca se debe demorar la administración de Ig a la espera de este. Según los hallazgos puede precisar coronariografía, angiotomografía computarizada (angio-TAC) o resonancia magnética (RM) cardiaca.
La afectación cardiológica en EK2:
- Miocarditis: casi universal (50-75%). Aparece antes de la afectación coronaria. Es transitoria con buena respuesta a la medicación antinflamatoria.
- Afectación coronaria: aparecen en el 25% de los pacientes no tratados, y en hasta el 5% de los tratados. Marca el pronóstico y en el 50% regresa a la normalidad en 2 años.
- Shock cardiogénico: en el 5% es la forma de debut. Suele asociar trombocitopenia y coagulopatía. Además, tiene más riesgo de resistencia a IgIV, afectación coronaria y disfunción miocárdica prolongada.
- Valvulitis con insuficiencia mitral: 25% en fase aguda. En relación con los marcadores de inflamación. No parece persistir en la evolución.
- Insuficiencia aórtica: 1%. Su presencia se asocia a dilatación coronaria.
- Pericarditis: 6-25%. Se asocia a fase aguda de enfermedad. Suele ser leve y transitoria.
Los siguientes hallazgos no son orientativos de EK: conjuntivitis exudativa, faringitis exudativa, úlceras orales, exantema vesicular, adenopatías generalizadas, esplenomegalia, leucopenia con linfocitosis o ausencia de trombocitosis y elevación de PCR y VSG más allá del 7 día de evolución2,6.
DIAGNÓSTICO DIFERENCIAL4,6
- Infección por virus: adenovirus, enterovirus, virus de Epstein-Barr, parvovirus B19, herpes 6, sarampión.
- Infecciones estafilocócicas o estreptocócicas mediadas por toxinas (escarlatina, síndrome del shock tóxico).
- Reacciones a fármacos (síndrome de Stevens Jonhson).
- Colagenopatías (inicio sistémico de artritis idiopática juvenil).
- Fiebre manchada de las montañas rocosas, otras riketsiosis o leptospirosis.
TRATAMIENTO EN FASE AGUDA
La aparición de aneurismas a nivel coronario aparece en un 25% de los casos en la evolución natural, el tratamiento adecuado en los 10 primeros días de evolución del cuadro (mejor en los 7 primeros), disminuye el riesgo de desarrollar aneurismas coronarios al 4% y de aneurismas gigantes al 1%.
La ausencia de diagnóstico y tratamiento adecuado pone al paciente en riesgo de muerte súbita por infarto o aneurisma o a padecer enfermedad coronaria asintomática que se diagnosticará al ser sintomática habitualmente en la edad adulta.
Factores de riesgo para desarrollar aneurismas coronarios5: el retraso diagnóstico, el retraso en el inicio del tratamiento, la ausencia de respuesta al tratamiento, las formas incompletas de presentación, las edades extremas (pacientes <12 meses y pacientes mayores) y puntuaciones elevadas en el score de Harada.
- El tratamiento de la fase aguda consiste en la administración de inmunoglobulinas IV (IgIV) a dosis de 2 g/kg en dosis única, en los primeros 10 días de evolución del cuadro (a ser posible antes del 7 día). Si el tratamiento se administra antes del 5 día de evolución de enfermedad, hay más riesgo de ser poco eficaz y de precisar una nueva administración. Se deben administrar más allá del día 10 en diagnósticos tardíos, si persiste elevación de los reactantes de fase aguda junto con afectación coronaria o persistencia de la fiebre.
- AAS a dosis antiinflamatorias (30-50 mg/kg/día, repartidos en 4 tomas al día), que se mantendrá hasta permanecer al menos 48-72 horas afebril. Pautándose luego a dosis antiagregantes (3-5 mg/kg/día en dosis única), que se mantendrá 6-8 semanas, suspendiéndose si la valoración cardiaca es normal y los reactantes de fase aguda y cifras de plaquetas normales2.
- El uso de corticoides coadyuvantes de primera línea es controvertido, pero cada vez hay más bibliografía que apoya su uso. Las guías japonesas de EK contemplan su uso como primera línea de tratamiento junto con IgIV y AAS en pacientes con puntuaciones de alto riesgo para resistencia inicial para las IgIV, describiendo acortamiento de los síntomas, disminución de la inflamación y mejor pronóstico coronario. En la actualidad fuera la población japonesa se puede considerar como terapia preventiva en EK graves, en EK con alto riesgo de resistencia a dosis inicial de IgIV y como terapia de rescate para pacientes que no responden inicialmente a las IgIV. No pudiéndose contemplar su uso de forma sistemática en todos los pacientes a día de hoy2,6.
Se considera fallo del tratamiento cuando la fiebre persiste o recurre tras 36 horas de la finalización de la administración de las IgIV, o no hay reducción del 50% del valor de la PCR. Esto ocurre en 10-20% de los pacientes. No hay evidencia en que tratamiento sería el más apropiado en este caso2. Expertos recomiendan administrar una segunda dosis de IgIV, responderían un 60-67% de los que no han respondido a la primera dosis de IgIV. Quedarían sin respuesta un 3-4%.
En estos pacientes también se han administrado corticoides en diferentes pautas:
- Metilprednisona IV a 2 mg/kg/día hasta desaparición de la fiebre y descenso de PCR. Después prednisolona/prednisona oral a 2 mg/kg/día hasta normalización PCR, con descenso y retirada progresiva en 2-3 semanas.
- Administración de bolos de metilprednisolona 30mg/kg/día durante 3 días. Continuando con metilprednisolona/prednisolona/prednisona IV/oral (según situación paciente) a 2 mg/kg/día, (se pasa a vía oral cuando desaparece fiebre y desciende PCR) hasta normalización PCR, descenso posterior hasta retirada en 2-3 semanas.
En pacientes refractarios a 2 dosis de IgIV o corticoides o afectación hemodinámica grave se pueden usar tratamientos biológicos:
- Infliximab: anticuerpo monoclonal anti TNF-alfa. Dosis de 6 mg/kg dosis única infundida en 2 horas. Puede repetirse a la semana.
- Anakinra: inhibidor de la acción de la IL-1. 2-6 mg/kg/día SC durante 15 días.
- Etanercept: proteína recombinante que bloquea la acción de TNF. 0,8 mg/kg/dosis IV semanal (3 dosis).
Anticoagulación:
- En caso de aneurismas gigantes, precisan anticoagulación asociando a las dosis antiagregantes de AAS anticoagulantes orales (warfarina o acenocumarol) o HBPM en dosis terapéuticas.
Al menos un 10-20% de los pacientes presentan a las 36 horas de la administración de IgIV persistencia o recidiva de la fiebre, se denomina resistencia a las Ig, teniendo estos más riesgo de aneurismas coronarios. Estos son los pacientes que tratan de identificar los diferentes Scores, pero que aún deben ser validados para poblaciones no japonesas 6.
Score de predicción de resistencia a IgIV de Kobayashi7: con una puntuación ≥5. Sensibilidad del 86%. Especidad del 68%. Validado para población japonesa.
- Sodio <133 mEq/l: 2 puntos.
- AST >100U/l: 2 puntos.
- Día de enfermedad al inicio tratamiento ≤4: 2 puntos8.
- Neutrófilos ≥80%: 2 puntos8.
- PCR ≥10 mg/dl: 1 punto8.
- Edad ≤12 meses: 1 punto 8.
- Plaquetas ≤300 000: 1 punto.
Para población no japonesa, para tratar de predecir la resistencia a IgIV:
- Edad <12 meses.
- Dilatación coronaria previa al inicio del tratamiento.
- EK que debuta con shock.
- EK con síndrome de activación macrofágico.
Todavía no hay un esquema para aplicar con suficiente evidencia en el caso de la resistencia a las IgIV, pudiéndose administrar:
- Una nueva dosis de IgIV a 2 g/kg.
- IgIV más corticoides (metilprednisolona IV a 2 kg/kg/día cada 8 horas hasta quedar afebril, seguido de prednisona a 2 mg/kg/día hasta normalización de PCR, con pauta de descenso oral durante 2-3 semanas)6.
- Infliximab.
La función miocárdica suele recuperarse al mejorar la inflamación y las manifestaciones sistémicas, tras la administración de las IgIV. En los casos en que se asocie shock e inestabilidad hemodinámica suele precisar asociar drogas vasopresoras y diuréticos.
Las principales complicaciones en la fase aguda pueden ser la rotura de un aneurisma coronario, la trombosis de un aneurisma o un infarto agudo de miocardio. Si se sospechan por deterioro función ventricular, dilatación progresiva coronaria, precisan de incrementar nivel de antiagregación (añadir clopidogrel al AAS).
SEGUIMIENTO POSTERIOR
En seguimiento una vez pasada la fase aguda (4-6 semanas) los pacientes se estratifican según la afectación coronaria que presenten en cualquier momento de la enfermedad en diferentes grupos de riesgo.
Si la valoración cardiológica a las 4-6 semanas es normal se puede suspender el tratamiento antiagregante y recibir alta cardiológica, presentado igual riesgo cardiológico que la población general. Si aparecen lesiones el tratamiento y seguimiento se debe individualizar según los hallazgos2,6.
Los pacientes que presentan aneurismas, estos se solucionan espontáneamente a los 3 meses el 15% y la mayoría a los 2 años, pero precisan seguimiento a largo plazo por riesgo de desarrollar estenosis en la zona del aneurisma.
Tras la administración de las IgIV deben demorarse durante 11 meses la administración de vacunas de virus vivos atenuados, por menor eficacia de estas. Si por un contacto se tuvieran que administrar, se debería revacunar al transcurrir los 11 meses6.
Mientras se tome AAS como antiagregante no se debe administrar ibuprofeno, pues antagoniza su efecto6.
Se recomienda la vacunación del gripe estacional a todos los pacientes >6 meses que han padecido un Kawasaki y toman AAS como antiagregante, así como a sus convivientes por el riesgo de presentar un síndrome de Reye6.
Síndrome del shock en la enfermedad de Kawasaki
En la fase aguda de la EK no suele presentarse inestabilidad hemodinámica salvo que se produzca un efecto adverso en la infusión de IGIV. Se define síndrome de shock asociado a EK en pacientes que precisan ingreso en UCI, perfusión de drogas vasoactivas o necesidad de expansión de volumen:
- Hipotensión arterial sistólica para la edad (1-28 días: <60 mmHg, 1-12 meses: <70 mmHg; niños de 1-10 años: < [70 + (2 × edad)] mmHg ; niños >10 años ≤90 mmHg.
- Caída de la TA sistólica ≥20 mmHg de la línea basal.
- Signos clínicos de hiperfusión tisular (taquicardia mantenida, enlentecimiento del tiempo de revascularización capilar, frialdad acra, oliguria o alteración del nivel conciencia no atribuible a otras causas).
Está más descrito en mujeres, pacientes con datos analíticos con mayor inflamación, con menor cifra de hemoglobina y plaquetas, disfunción cardiaca sistólica y diastólica, mayor tasa de resistencia a las IgIV y mayor tasa de aneurismas coronarios. La función cardiovascular se restablece, permaneciendo la disfunción cardiaca diastólica durante la fase crónica del seguimiento y en un 5% de los pacientes puede ser la forma de debut6.
Síndrome inflamatorio multisistémico relacionado con la infección SARS-CoV-2
Durante la pandemia mundial del SARS-CoV-2 los niños menores de 18 años parecen tener unas tasas de infección por el nuevo coronavirus mucho más bajas que los adultos, lo que no se sabe si representa una menor susceptibilidad a la infección o una mayor tasa de infección asintomática. Y unos cuadros mucho menos graves que los adultos.
Entre los diferentes países se ha demostrado que las tasas de infección severa o fallecimiento en niños por COVID-19 son bajas. A principios de mayo de 2020, en los picos pándemicos de la COVID-19 se reportaron en pacientes pediátricos casos de un síndrome inflamatorio sistémico de expresividad variable. Este cuadro comparte datos clínicos y analíticos con la EK, el síndrome del shock tóxico o los síndromes de activación macrofágica. Pudiendo evolucionar de forma grave con miocarditis y shock cardiogénico. Este cuadro ha tratado de ser definido sin llegar a un acuerdo de su definición y manejo por diferentes organismos como la OMS, la CDC o el RCPCH.
Este nuevo cuadro ha coincido temporalmente con la pandemia por SARS-COV-2, presentado la mayoría de los pacientes infección activa o reciente por dicho virus. La mayoría presenta serologías IgG positivas y elevados marcadores de infección y a pesar de no estar clara la relación etiológica, parece que se debe a una desregulación de la respuesta inmunitaria desencadenada por el virus, más que a una acción patógena directa del mismo.
Un 78% presentaban evidencia de infección previa o activa por SARS-CoV-2, todos presentaban fiebre y síntomas inespecíficos como dolor abdominal, vómitos, diarrea, exantema o conjuntivitis, con afectación cardiaca y renal. Marcadores bioquímicos de inflamación (elevación PCR y ferritina), datos bioquímicos de fallo cardiaco y un 50% criterios clínicos de enfermedad de Kawasaki. Además puede asociar linfopenia, coagulopatía, hiponatremia, hipoalbuminemia, y elevación de enzimas cardiacas, LDH y ferritina9. Siendo este grupo de pacientes algo mayores que en la enfermedad de Kawasaki y con marcadores de infección algo más elevados10. Se presenta en niños con una edad media de 9 años, ligeramente más frecuente en varones, de etnia no caucásica y con sobrepeso. Se han presentado 3 patrones clínicos
- Cuadro clínico con fiebre o dolor abdominal o exantema o conjuntivitis e importa elevación reactantes fase aguda o enzimas cardiacas.
- Cuadro clínico compatible con miocarditis (disfunción VI y elevación pro-BNP), shock séptico o tóxico.
- Cuadro clínico compatible con Kawasaki completo/incompleto.
Suelen responder bien a tratamiento con IgIV, pero hasta un 20% ha precisado tratamiento antiinflamatorio adicional9,11-16.
El principal tratamiento recibido fueron inmunoglobulinas IV, corticoides y soporte hemodinámico. Se pueden beneficiar de agentes anti-IL112,17.
CUADERNO DEL PEDIATRA
- Es fundamental un alto índice de sospecha de la enfermedad de Kawasaki, por ello se debe tener siempre en mente. Ante cuadros febriles de más de 5 días sin explicación, se debe vigilar la aparición de síntomas clásicos.
- En caso de sospecha asegurarse de una adecuada valoración clínica, incluyendo analítica y estudio cardiológico, para instaurar si procede el tratamiento con IgIV y AAS antes de los 10 días de evolución de enfermedad.
- En pacientes tratados con IgIV se deben demorar la administración de vacunas de virus vivos atenuados durante 11 meses tras la administración de las inmunoglobulinas. Si por un contacto se debiera vacunar antes, revacunar pasados esos 11 meses.
- Mientras se esté en tratamiento antiagregante con AAS se debe evitar la toma de ibuprofeno. Y se debe vacunar de la gripe al paciente y sus convivientes, por el riesgo de síndrome de Reye (se debe sustituir el AAS por otro antiagregante tras la vacunación).
- En todo paciente pediátrico que haya pasado la infección por COVID-19, se debe estar al tanto si en las siguientes semanas comienza con cuadro febril prolongado, asociado a síntomas de enfermedad de Kawasaki, clínica digestiva o síntomas de shock, que se pueda tratar de un cuadro de síndrome inflamatorio multisistémico, para su derivación a un centro con cuidados intensivos y si procede un tratamiento precoz.
BIBLIOGRAFÍA
- Younger DS. Epidemiology of the Vasculitides. Neurol Clin. 2019 May;37(2):201-217.
- Barrios Tascón A, Centeno Malfaz F, Rojo Sombrero H, Fernández-Cooke E, Sánchez-Manubens J, Pérez-Lescure Picarzo J, et al. Consenso nacional sobre diagnóstico, tratamiento y seguimiento cardiológico de la enfermedad de Kawasaki. An Pediatr (Barc). 2018;89:188.e1-188.e22.
- Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis J. 2010;29:483-8.
- Sociedad Argentina de Pediatría, Sociedad Argentina de Cardiología. Enfermedad de Kawasaki: Consenso interdisciplinario e intersociedades. Arch Argent Pediatr. 2016;114:1-18.
- Duignan S, Doyle SL, McMahon CJ. Refractory Kawasaki disease: diagnostic and management challenges. Pediatric Health Med Ther. 2019;10:131-9.
- McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation. 2017;135:e927-e999.
- Kobayashi T, Inoue Y, Takeuchi K, Okada Y, Tamura K, Tomomasa T, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113:2606-12.
- Egami K, Muta H, Ishii M, Suda K, Sugahara Y, Iemura M, Matsuishi T. Prediction of resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease. J Pediatr. 2006;149:237-40.
- Toubiana J, Poirault C, Corsia A, Bajolle F, Fourgeaud J, Angoulvant F, et al. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. BMJ. 2020 Jun 3;369:m2094.
- Whittaker E, Bamford A, Kenny J, Kaforou M, Jones CE, Shah P, et al. Clinical Characteristics of 58 Children With a Pediatric Inflammatory Multisystem Syndrome Temporally Associated With SARS-CoV-2. JAMA. 2020 Jul 21;324:259-69.
- Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395:1607-8.
- Koné-Paut I, Cimaz R. Is it Kawasaki shock syndrome, Kawasaki-like disease or pediatric inflammatory multisystem disease? The importance of semantic in the era of COVID-19 pandemic. RMD Open. 2020;6:e001333.
- Pouletty M, Borocco C, Ouldali N, Caseris M, Basmaci R, Lachaume N, et al. Paediatric multisystem inflammatory syndrome temporally associated with SARS-CoV-2 mimicking Kawasaki disease (Kawa-COVID-19): a multicentre cohort. Ann Rheum Dis. 2020;79:999-1006.
- Harahsheh AS, Dahdah N, Newburger JW, Portman MA, Piram M, Tulloh R, McCrindle BW, de Ferranti SD, Cimaz R, Truong DT, Burns JC. Missed or delayed diagnosis of Kawasaki disease during the 2019 novel coronavirus disease (COVID-19) pandemic. J Pediatr. 2020;222:261-2.
- Belot A, Antona D, Renolleau S, Javouhey E, Hentgen V, Angoulvant F, et al. SARS-CoV-2-related paediatric inflammatory multisystem syndrome, an epidemiological study, France, 1 March to 17 May 2020. Euro Surveill. 2020;25:2001010.
- Chiotos K, Bassiri H, Behrens EM, Blatz AM, Chang J, Diorio C, et al. Multisystem Inflammatory Syndrome in Children During the Coronavirus 2019 Pandemic: A Case Series. J Pediatric Infect Dis Soc. 2020;9:393-8.
- Grupo de redacción multidisciplinar conformado por la Asociación Española de Pediatría, et al. Consenso nacional sobre diagnóstico, estabilización y tratamiento del Síndrome Inflamatorio Multisistémico Pediátrico vinculado a SARS-CoV-2 (SIM-PedS). En: SEIP [en línea] [consultado el 17/03/2021]. Disponible en: www.seipweb.es/wp-content/uploads/2020/08/2020.-Consenso-nacional-sobre-diagnóstico-estabilización-y-tratamiento-del-Síndrome-Inflamatorio-Multisistémico-Pediátrico-vinculado-a-SARS-CoV-2-SIM-PedS.pdf