

Hematomas en el lactante, ¿podría ser hemofilia?
2 Pediatra. CS Zona VIII. Albacete. (España).
PUNTOS CLAVE
- Los hematomas postraumáticos en miembros inferiores son frecuentes en los lactantes durante la época de inicio del gateo y la deambulación.
- En la mayoría de las ocasiones, la anamnesis y la exploración física serán suficientes para descartar signos de alarma, como la localización inusual de estos.
- Si los hematomas aparecen en un lactante menor de 6 meses debemos descartar la existencia de malos tratos.
- Cuando encontremos signos de alarma, como hematomas en localizaciones inusuales o sangrados excesivos y/o prolongados, solicitaremos pruebas para descartar alteraciones de la hemostasia.
CASO CLÍNICO
Lactante varón de 14 meses que consulta por hematomas y sangrados mucocutáneos prolongados tras traumatismos leves. Como antecedentes personales, consta que fue un recién nacido a término de peso adecuado para la edad gestacional, correctamente vacunado, sin medicación crónica, con antecedentes de frenectomía sublingual con electrobisturí al mes de vida sin incidencias. Presenta antecedentes familiares de coagulopatía: la madre presentó hemorragia puerperal leve secundaria a atonía uterina que cede con tratamiento médico, abuela paterna diagnosticada de trombopatía leve por defecto de liberación de gránulos densos, y prima materna con déficit de factor VII.
Refieren inicio de la clínica a los 7 meses de vida, coincidiendo con el comienzo del “gateo”, con aparición de hematomas en tórax y miembros (Figuras 1 y 2). A los 9 meses presenta sangrado de una pequeña erosión perianal en relación con su estreñimiento. A los 13 meses consulta por hematoma subungueal postraumático en primer dedo de pie izquierdo que motiva cuatro visitas a Urgencias por persistencia del sangrado, donde se aplicaron medidas para favorecer la hemostasia.
Figura 1. Hematoma en tórax. Mostrar/ocultar
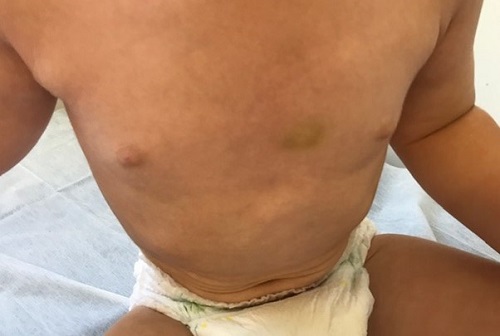
Figura 2. Hematomas en extremidad inferior. Mostrar/ocultar
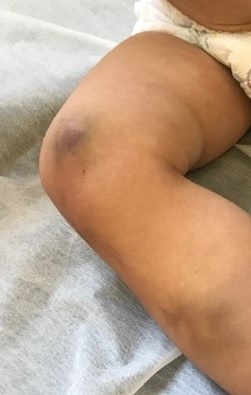
Ante la clínica que presenta y los antecedentes familiares, se solicita un estudio inicial de hemostasia con hemograma y coagulación, donde se objetiva un tiempo de tromboplastina parcial activado (TTPA) prolongado, 53,8” (valor normal: 23,4”-35”), sin otras alteraciones. Ante dicho resultado, se deriva al servicio de hematología para descartar un trastorno de la coagulación. En dicha consulta se repite el estudio, con datos similares al previo (TTPA: 50,2”), por lo que se solicita el nivel de actividad de los siguientes factores: factor de Von Willebrand (FVW): 75%, factor VIII: 155,8%, factor IX: 7,2%, factor XI: 102,2%, factor XII: 53%. Confirmado posteriormente con una nueva determinación: factor IX: 7,5%. Con todos estos datos, el paciente fue diagnosticado de hemofilia B leve. Para completar su estudio, se solicita estudio genético, donde se halla una mutación patológica en el gen F9, variante c.328G>C. Esta misma alteración genética fue descrita en la madre posteriormente tras realizar dicho estudio.
HEMOFILIA B
La hemofilia es un trastorno hereditario de la coagulación causado por el déficit o ausencia de un factor de coagulación. Podemos diferenciar tres tipos de hemofilia (A, B o C) según el factor involucrado1. Sin embargo, la clínica que presentan es similar debido a que estos factores actúan en la vía intrínseca de la coagulación1,2.
La hemofilia B se origina por la afectación del factor IX (F IX). Presenta un patrón de herencia recesiva ligada al cromosoma X, aunque también podemos encontrar mutaciones de novo. Se estima una incidencia estimada de 1/30 000 individuos, con afectación principalmente en el sexo masculino, pero sin distinción de etnias1-3.
Se debe conocer la gravedad que presenta, ya que influirá en la presentación clínica, evolución y pronóstico. Para poder determinar la gravedad, es necesario conocer la actividad del factor implicado en la hemofilia. Se considera una deficiencia grave si la actividad es inferior al 1% (<0,01 UI/ml), moderada si se encuentra entre 1-5% (≥0,01 y ≤0,05 UI/ml), o afectación leve si la actividad es superior al 5% pero inferior al 50% (≥0,05 y <0,40 UI/ml)1,2. Por otro lado, también se puede definir que un paciente presenta una hemofilia leve si, a pesar de tener una actividad del factor igual o superior al 40%, presenta clínica compatible y comparte una variante genética del factor implicado con un miembro de la familia afecto de hemofilia1.
Las manifestaciones clínicas dependen de la edad de aparición y su gravedad está determinada por la actividad del factor. Los pacientes con hemofilia grave tienen más probabilidades de presentar un sangrado espontáneo o iniciar la clínica desde una edad más temprana, incluso desde el nacimiento. Pueden presentar con mayor probabilidad sangrados abundantes tras un procedimiento convencional, como una venopunción, realización de prueba del talón, y en un pequeño porcentaje, pero potencialmente mortal, una hemorragia intracraneal. Sin embargo, los pacientes con enfermedad leve suelen presentar sangrado tras un traumatismo significativo o cirugías, incluso puede presentarse de forma diferida, o presentar clínica a una edad avanzada1-3.
En la infancia o adolescencia se presentan las manifestaciones clínicas más frecuentes, caracterizadas por hemorragias articulares (hemartrosis). El tobillo, la rodilla y el codo suelen ser las localizaciones más frecuentes. Las hemorragias musculares afectan principalmente a grandes grupos musculares, como cuádriceps o iliopsoas. En referencia a las hemartrosis, se manifiestan con dolor, tumefacción y aumento de la temperatura e impotencia funcional de la articulación afectada. Deben tratarse de forma precoz para evitar un daño irreparable, dado que pueden degenerar en una artropatía hemofílica. De forma similar hay que actuar con las hemorragias musculares, dado que su evolución tórpida puede degenerar en un síndrome compartimental o un pseudotumor1.
Otras manifestaciones que también se pueden encontrar son los sangrados a nivel nasofaríngeo, como epistaxis de repetición, sangrado a nivel oral de forma espontánea o producido tras un traumatismo menor o cirugía. Menos frecuente sería el sangrado gastrointestinal o genitourinario, con hematemesis o hematuria de causa desconocida o recidivante1.
DIAGNÓSTICO
Cuando se sospecha una alteración en la coagulación es fundamental realizar una anamnesis orientada, prestando especial atención a los antecedentes familiares y a la presencia de clínica hemorrágica: comienzo, duración, episodios previos, localización, circunstancias acompañantes, infecciones, fármacos y enfermedades subyacentes. El sangrado visible en piel o mucosas ofrece una importante información sobre la presencia de una coagulopatía. Es característico el sangrado espontáneo, es decir, una hemorragia sin una causa externa identificable o una hemorragia excesiva tras una lesión o traumatismo menor. Ante la presencia de hematomas y sangrados postraumáticos es necesario descartar malos tratos. Los pediatras de Atención Primaria contamos con el conocimiento de la familia y con el seguimiento evolutivo, que nos aportará datos muy valiosos para poder detectarlos.
Ante una historia de sangrado inusual o en pacientes asintomáticos con antecedentes familiares de coagulopatía se deben solicitar las cuatro pruebas básicas para valorar la hemostasia: el recuento plaquetar que aporta el hemograma, el tiempo de hemorragia (TH), el tiempo de protrombina (TP) y el tiempo de tromboplastina parcial activado (TTPA)4. Estas dos últimas determinaciones exploran la hemostasia secundaria, se expresan en segundos y se comparan con un tiempo control que define cada laboratorio. Cuando estos tiempos están alargados por encima del tiempo control, debe hacerse una segunda determinación para confirmarlo, ya que en aproximadamente un 50% de los casos no se confirma ninguna alteración obedeciendo a la interferencia de factores preanalíticos.
La hemofilia se caracteriza por presentar un TTPA prologando, que incluso podría encontrarse en límite normal en caso de pacientes con hemofilia leve1,2. El tiempo de protrombina, tiempo de hemostasia y el nivel de plaquetas son normales en esta enfermedad; en caso de encontrarse alterado, deberemos sospechar otra etiología4. En la Tabla 1 se exponen los diagnósticos asociados a las diferentes alteraciones del estudio de la coagulación4.
Tabla 1. Diagnóstico diferencial según alteración en el estudio de coagulación. Mostrar/ocultar
Para completar el estudio, se realizará la determinación del nivel de actividad del factor o factores que se sospechen estén disminuidos1,2. Dicho estudio podrá ser dirigido si el paciente presenta antecedentes familiares de hemofilia y se conoce con exactitud el factor implicado; o se realizará un estudio de todos los factores si es un caso nuevo. El valor de la actividad nos confirmará la enfermedad en caso de encontrarse alterado.
Se deben interpretar los resultados de forma correcta, y saber que diversas situaciones no relacionadas directamente con la enfermedad pueden variar los resultados. Por ello, se precisan dos determinaciones analíticas con un nivel de actividad del factor inferior al normal para poder confirmar la enfermedad.
En algunas ocasiones, se puede completar el estudio de hemofilia con la realización de pruebas moleculares o genéticas. Es un estudio complejo, dado que se han descrito multitud variantes patogénicas, por ejemplo, deleciones, duplicaciones o mutaciones en el gen F8 en la hemofilia A, o gen F9 en caso de la hemofilia B5. La genética tiene implicaciones en la gravedad de la enfermedad, dado que se han descrito variantes patogénicas relacionas con una afectación grave de la enfermedad. Por otro lado, ayuda a predecir el riesgo de formación de inhibidores, autoanticuerpos que se forman tras la administración del factor exógeno, que interfieren con la función del factor. Este estudio también contribuye a confirmar personas portadoras de la enfermedad y así realizar un seguimiento estrecho de esta enfermedad y ofrecer un asesoramiento y atención integral. Por último, permite realizar un consejo genético individualizado y asesoramiento previo a la concepción, o un diagnóstico prenatal, por biopsia de vellosidad coriónica o amniocentesis, según la edad gestacional1,4,5.
ACTITUD TERAPÉUTICA
El manejo óptimo de las personas diagnosticadas de hemofilia es complejo y el objetivo debe ir encaminado al diagnóstico precoz, manejo integral, tratamiento temprano y prevención del desarrollo de complicaciones. La educación sobre la enfermedad al paciente y cuidadores es fundamental.
Educación y prevención
En la consulta de atención primaria se debe realizar el seguimiento de esta patología y ofrecer la información necesaria para conseguir un estilo de vida saludable, evitando el desarrollo de complicaciones. La higiene y cuidado dental adecuados son esenciales para prevenir enfermedades dentales y/o gingivales que aumenten el riesgo de hemorragia. Es importante la realización de ejercicio físico de forma regular, evitando aquellos deportes de impacto/contacto, y así prevenir el sobrepeso/obesidad y generar efectos beneficiosos a nivel locomotor. Se deben dar pautas de actuación en caso de traumatismos o algias diversas, dado que es importante conocer aquella medicación que se debe evitar por riesgo de hemorragia, como son los antiinflamatorios no esteroideos2,6.
Hay que conocer que la administración de vacunas deberá realizarse por vía convencional con una aguja del menor calibre posible, con posterior aplicación de medidas locales durante unos minutos tras la punción; o por vía subcutánea2,6. Además, es importante recomendar la vacunación frente a la hepatitis A, puesto que, a pesar de los grandes avances en la investigación, continúa existiendo un riesgo en la transmisión de infecciones derivado de la infusión del plasma.
Tratamiento
A nivel terapéutico y/o profiláctico, el tratamiento principal de la hemofilia es el empleo de concentrado plasmático recombinante2,3,6. La actuación ante un sangrado menor, tal como epistaxis, mínimo sangrado cutáneo, se basa en medidas locales, como aplicación de hielo, presión o elevación. Si presenta una evolución tórpida, las terapias tópicas con agente antifibrinolíticos, como ácido tranexámico, también pueden resultar útiles6.
Sin embargo, ante un paciente con hemofilia y una hemorragia aguda, el objetivo será aumentar el nivel de actividad del factor hasta un valor que permita lograr la hemostasia. Dicho valor dependerá del origen y gravedad de la hemorragia y la vida media del concentrado utilizado. Una unidad de concentrado de F IX por kg de peso sube un 1% la actividad del F IX; y su vida media es de 18-24 horas (Tabla 2)2,6,7.
Tabla 2. Manejo de actuación ante hemofilia B. Mostrar/ocultar
El manejo profiláctico de la hemofilia se realizará solo en aquellos pacientes que presentan una enfermedad grave, dado que el uso de forma rutinaria de este concentrado puede conllevar el desarrollo de inhibidores. En el caso de la hemofilia B, puede ocurrir en un 5% hasta un 15% de los casos2,6.
Finalmente, ante un paciente con dicha enfermedad que requiera una cirugía, preferiblemente se realizará de forma programada; con un plan integral de manejo, una adecuada comunicación paciente-familiares/facultativos, y tras realizar un estudio completo, para identificar posibles problemas asociados, uso de medicación e incluso cribado de inhibidores6,7.
BIBLIOGRAFÍA
- Hoots WK, Shapiro AD. Clinical manifestations and diagnosis of hemophilia. En: UpToDate. Leung LK (Ed), Uptodate, Waltham MA [en línea] [consultado el 04/04/2023]. Disponible en: www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-hemophilia?search=Clinical%20manifestations%20and%20diagnosis%20of%20hemophilia
- Cervera Bravo, A. Fisiopatología y trastornos de la coagulación hereditarios más frecuentes. Pediatr Integral. 2012;XVI:387-98.
- García Sánchez P, Martín Sánchez J, Rivas Pollmar MI, Álvarez Román MT, Jiménez Yuste V. Hemofilia: naturaleza de las visitas a urgencias pediátricas. An Pediatr (Barc). 2019;91:394-400.
- Moreno Jiménez G, Zamora Gómez M. Hemograma y estudio de coagulación. Form Act Pediatr Aten Prim. 2008;1:95-100.
- Hoots WK, Shapiro AD, Heiman M. Genetics of hemophilia A and B. En: UpToDate. Leung LK (Ed), Uptodate, Waltham MA [en línea] [consultado el 04/04/2023]. Disponible en: www.uptodate.com/contents/genetics-of-hemophilia-a-and-b?search=Genetics%20of%20hemophilia%20A%20and%20B
- Hoots WK, Shapiro AD. Hemophilia A and B: routine management including prophylaxis. En: UpToDate. Leung LK (Ed), Uptodate, Waltham MA [en línea] [consultado el 04/04/2023]. Disponible en: www.uptodate.com/contents/hemophilia-a-and-b-routine-management-including-prophylaxis?search=Hemophilia%20A%20and%20B:%20routine%20management%20including%20prophilaxis
- Hoots WK, Shapiro AD. Treatment of bleeding and perioperative management in hemophilia A and B. En: UpToDate. Leung LK (Ed), Uptodate, Waltham MA [en línea] [consultado el 04/04/2023]. Disponible en: www.uptodate.com/contents/treatment-of-bleeding-and-perioperative-management-in-hemophilia-a-and-b?search=Treatment%20of%20bleeding%20and%20perioperative%20management%20in%20hemophilia%20A%20and%20B
Lecturas recomendadas
- Cervera Bravo, A. Fisiopatología y trastornos de la coagulación hereditarios más frecuentes. Pediatr Integral. 2012;XVI:387-98.
Se trata de un artículo muy completo que ofrece información sobre los principales trastornos de la coagulación en pediatría, de forma concisa, pero dirigida a ayudar en su diagnóstico y tratamiento.
- Hoots WK, Shapiro AD. Clinical manifestations and diagnosis of hemophilia. En: UpToDate. Leung LK (Ed), Uptodate, Waltham MA. [en línea] [consultado el 04/04/2023]. Disponible en: www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-hemophilia?search=Clinical%20manifestations%20and%20diagnosis%20of%20hemophilia
En este artículo se realiza una revisión detallada y actualizada sobre la epidemiología de la hemofilia, las diversas manifestaciones clínicas que puede presentar dividido por aparatos, las diferencias según la edad a la que se manifiesta la enfermedad, especificando los diversos tipos de sangrado que puede presentar, así como sus complicaciones. Por otro lado, informa de forma precisa sobre el diagnóstico de la enfermedad, incluido el estudio genético.
- Hoots WK, Shapiro AD. Hemophilia A and B: routine management including prophylaxis. En: UpToDate. Leung LK (Ed), Uptodate, Waltham MA [en línea] [consultado el 04/04/2023]. Disponible en: www.uptodate.com/contents/hemophilia-a-and-b-routine-management-including-prophylaxis?search=Hemophilia%20A%20and%20B:%20routine%20management%20including%20prophilaxis
Este artículo ofrece una información precisa sobre los diversos tratamientos y formas de administración en pacientes con hemofilia, así como pautas para profilaxis primaria y secundaria de la enfermedad.