

Lactante con deformación craneal
2 Pediatra. Tutora de MIR de pediatría. CS Delicias. Málaga (España).
RESUMEN: PUNTOS CLAVE PARA EL PEDIATRA DE ATENCIÓN PRIMARIA
SUMMARY: KEY POINTS FOR PRIMARY CARE PEDIATRICIANS
PUNTOS CLAVE
- La craneosinostosis, o cierre prematuro de una o más suturas craneales, constituye una entidad poco frecuente. El pediatra de Atención Primaria desempeña una labor importante en su diagnóstico precoz.
- El diagnóstico es fundamentalmente clínico, aunque puede apoyarse en la radiografía de cráneo.
- Debemos diferenciar la plagiocefalia postural, en aumento las últimas décadas, de la secundaria a craneosinostosis.
- Su reconocimiento y derivación precoz al especialista en Neurocirugía es extremadamente importante.
- Hay que realizar una adecuada exploración física en busca de alteraciones del desarrollo motor y cognitivo.
- El tratamiento quirúrgico clásico está siendo reemplazado en los últimos años por cirugía endoscópica mínimamente invasiva.
INTRODUCCIÓN
La craneosinostosis consiste en el cierre precoz de una o más suturas craneales; en el periodo neonatal se manifiesta como la alteración de la forma del cráneo, cuyo crecimiento se ve reducido en dirección perpendicular a la sutura afecta1. Hablamos de craneosinostosis aislada o múltiple según estén afectadas una o varias suturas craneales. La craneosinostosis puede ser primaria, secundaria o formar parte de un síndrome.
Se desconoce la etiología de la craneosinostosis primaria, aunque se ha postulado que participen factores como el descenso precoz de la cabeza fetal, la presentación de nalgas prolongada, la posición fetal anormal, la existencia de múltiples fetos en la cavidad uterina, el oligohidramnios y el feto macrosómico. Se han identificado mutaciones genéticas hasta en un 20% de los casos2.
Las craneosinostosis sindrómicas suelen ser múltiples, identificándose mutaciones genéticas con herencia autosómica dominante o mutaciones de novo. Se dividen en cuatro grupos: craneosinostosis aisladas, craneosinostosis con sindactilia, craneosinostosis con polidactilia y sindactilia, y craneosinostosis con otros hallazgos somáticos.
La craneosinostosis secundaria constituye la forma más frecuente de sinostosis. Puede ser secundaria a enfermedades hematológicas (anemia de células falciformes), a trastornos endocrinos (hipertiroidismo, hipofosfatemia, hiperalcemia) o a microcefalia. En la microcefalia disminuyen las fuerzas de distensión sobre las suturas, lo que condicionará la osificación y el cierre prematuro de estas sin que aparezcan asimetrías o rebordes óseos. En estos casos, la presión intracraneal es normal y la fusión prematura de las suturas no compromete el desarrollo del cerebro, por lo que no se requiere ninguna intervención terapéutica o quirúrgica.
CASO CLÍNICO
Lactante de dos meses que es llevado a la consulta de Pediatría de Atención Primaria por presentar un cuadro catarral de dos días de evolución.
Entre sus antecedentes personales figuran embarazo controlado, test de Apgar 9/10, edad gestacional 39 semanas, peso al nacer 3150 g, talla 52 cm, pruebas metabólicas normales y screening de hipoacusia normal.
En la exploración física destaca una cabeza larga y estrecha (cráneo dolicocefálico) con protusión frontal, fontanela anterior abierta y posterior cerrada, y un perímetro cefálico de 42 cm (p97). En la palpación del cráneo se objetiva una sutura sagital inexistente, sin palpación de reborde óseo. El resto de la exploración física no demuestra alteraciones. Desarrollo psicomotor normal para su edad.
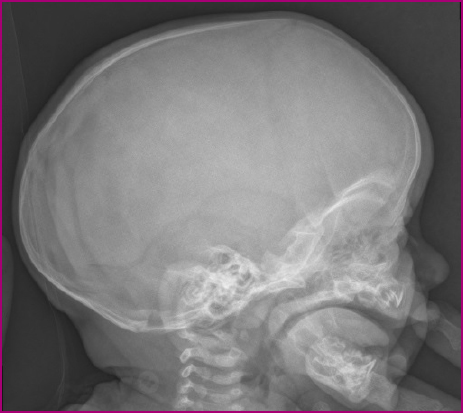
Ante la sospecha de craneosinostosis se realiza una radiografía de cráneo (Figura 1), confirmándose la fusión de la sutura sagital. En este momento se le deriva al especialista en Neurocirugía Pediátrica.
Figura 1. Radiografía lateral de cráneo del paciente Mostrar/ocultar
FISIOPATOLOGÍA
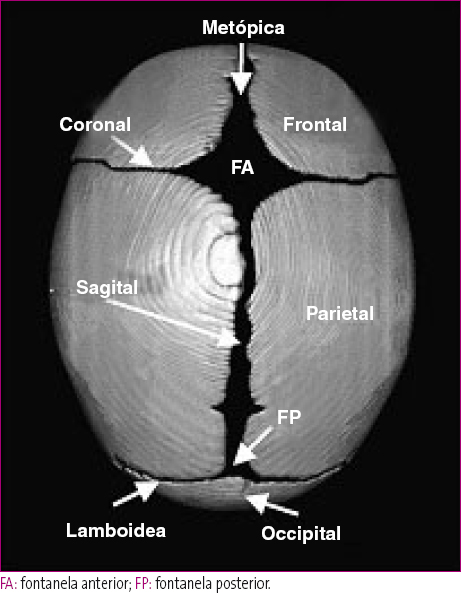
La osificación de la bóveda craneal se inicia en la región central de cada hueso y se extiende hacia las suturas craneales. Hay cuatro suturas mayores: coronal (separa los dos huesos frontales del hueso parietal), metópica (separa ambos huesos frontales), sagital (separa los dos huesos parietales) y lambdoidea (separa los huesos parietales del occipital). Las suturas coronal, metópica y sagital delimitan la fontanela anterior, mientras que la fontanela posterior se forma por la intersección de la sutura sagital y las lambdoideas (Figura 2)3.
Figura 2. Suturas craneales3 Mostrar/ocultar
La sinostosis precoz de cada una de estas suturas originará las diferentes alteraciones en la conformación del cráneo (Figura 3)4.
Figura 3. Diferentes alteraciones en la conformación del cráneo6 Mostrar/ocultar
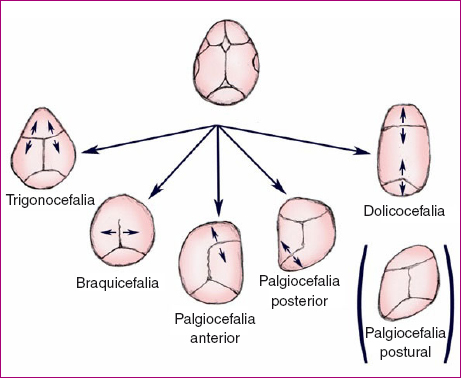
- Cabeza estrecha y excesivamente larga: dolicocefalia o escafocefalia. Constituye la forma más frecuente de craneosinostosis primaria, consecuencia de sinostosis sagital. Predomina en varones. Puede asociar abombamiento frontal y occipital incrementando la alteración de la forma craneal. Hay que diferenciar la dolicocefalia sinostótica de la posicional (lactantes hipotónicos o neonatos pretérmino que descansan apoyados sobre la zona lateral del cráneo la mayor parte del tiempo).
-
Cabeza triangular con la frente en forma de quilla: trigonocefalia secundaria a la sinostosis de la sutura metópica. Si incluye la sutura frontonasal aparece hipotelorismo. Puede asociar holoprosencefalia, agenesia del cuerpo calloso, meningocele, fisura palatina, coloboma y retraso mental, así como hipertensión intracraneal. Se ha visto que un tercio de los niños con trigonocefalia desarrollan trastornos cognitivos y de conducta. Puede observarse en:
- El síndrome de Opitz-C: estrechez bifrontal, nariz ancha y corta, microcórnea, micrognatia y polidactilia. Evoluciona a la muerte en el primer año de vida.
- El síndrome de Baller-Gerold: aplasia de radio, retraso del crecimiento, ausencia de huesos pequeños de la mano, ano imperforado y retraso cognitivo en un 50% de los casos.
- Síndrome por hidantoína fetal: crecimiento retardado, fontanela anterior amplia, hipertelorismo, labio leporino y fisura palatina, cuello corto, hipoplasia ungueal y de falanges distales y trastorno cognitivo variable.
- Deleción del brazo corto del cromosoma 9: hipoplasia medio-facial, narinas antevertidas, alteraciones auriculares, pliegues de flexión digital extras, cardiopatía y trastorno cognitivo grave.
- Cabeza ancha: braquicefalia. Sinostosis bicoronal o bilambdoidea, normalmente sindrómica. Si el crecimiento es más marcado hacia arriba se denomina turricefalia. En el lactante, la principal causa no es sinostótica, sino por la posición en decúbito supino prolongada. Esta deformación, con plagiocefalia o sin ella, ha aumentado tras la recomendación de dormir en decúbito supino como prevención del síndrome de muerte súbita del lactante.
-
Cabeza asimétrica: plagiocefalia (cabeza oblicua). Puede ser anterior o posterior.
La plagiocefalia anterior se presenta tras el cierre prematuro de una sutura coronal. Es más frecuente en mujeres y en el lado derecho, y comprende el 20% de las sinostosis primarias.
La plagiocefalia posterior puede ser consecuencia de sinostosis de la sutura lambdoidea o, en la mayoría de los casos, por moldeamiento posicional. La recomendación de la Asociación Americana de Pediatría de dormir en decúbito supino para la prevención de la muerte súbita ha ocasionado un incremento de esta patología en las últimas décadas5. Este moldeamiento podría comenzar intraútero, exacerbándose postnatalmente cuando el niño descansa sobre el occipital aplanado. Puede existir alopecia por presión en el lado afecto. En la plagiocefalia con sinostosis, el abombamiento frontal y parietal contralateral son más acusados, y la oreja ipsilateral al lado aplanado puede desplazarse en dirección posterioinferior. - Cabeza en trébol: oxicefalia. Cierre prematuro de todas las suturas a excepción de la sagital. Asocia graves alteraciones faciales y presenta generalmente hipertensión intracraneal y retraso mental.
ABORDAJE EN ATENCIÓN PRIMARIA
El examen clínico es la clave para el diagnóstico6. La observación de la forma, la palpación de un reborde óseo a lo largo de la sutura, así como la aplicación de una presión firme alternante con los pulgares sobre ambos lados de la sutura con el objetivo de causar un relativo desplazamiento de los huesos vinculados por esta, permite llegar al diagnóstico de craneosinostosis.
En la exploración física puede ser útil conocer el índice cefálico (IC), cociente entre el ancho de la cabeza (distancia entre las eminencias parietales) y el largo en la dimensión anteroposterior (IC = ancho / largo x 100). Un IC <76% indica cráneo largo y estrecho, mientras que un IC >81% señala un cráneo ancho y acortado anteroposteriormente.
En la radiografía de cráneo (Figura 1) podemos observar una línea de esclerosis y engrosamiento del hueso adyacente a la sinostosis. La tomografía computarizada es el procedimiento de elección para determinar la extensión de la sinostosis, así como para descartar alteraciones estructurales en el parénquima cerebral7.
TRATAMIENTO
La cirugía está indicada en casos sintomáticos (cefalea, vómitos o cambios en la visión) secundarios a un aumento de la presión intracraneal, o para la corrección de la deformidad. Si a los dos meses de vida la forma craneal no ha mejorado, es improbable que se resuelva con la edad. No está indicada la corrección quirúrgica en los casos de plagiocefalia postural. En estos casos, la gran mayoría de los niños mejora con maniobra de reposicionamiento y terapia física, aunque en casos graves pueden usarse cascos de moldeado. La derivación precoz es muy importante en aquellos niños que pueden beneficiarse de una cirugía endoscópica, la cual proporciona una excelente alternativa quirúrgica, al ser mínimamente invasiva, con una media de estancia hospitalaria de un día y mínima pérdida sanguínea8. Además, diversos estudios señalan que los resultados son mejores cuando se realiza la cirugía en lactantes menores de seis meses.
Tras la corrección quirúrgica, debemos realizar un seguimiento estrecho para detectar resinostosis, así como signos y síntomas de hipertensión intracraneal en los niños con factores de riesgo8,9.
BIBLIOGRAFÍA
- Slater BJ, Lenton KA, Kwan MD, Gupta MD, Wan DC, Longaker MT. Cranial sutures: a brief review. Plast Reconstr Surg. 2008;121:170-8.
- Johnson D, Wilkie AO. Craniosynostosis. Eur J Hum Genet. 2011;19:369-76.
- Cunningham ML, Heike CL. Evaluation of the infant with an abnormal skull shape. Curr Opin Pediatr. 2007;19:645-51.
- Sheth RD, Ranalli N, Aldana P. Pediatric Craniosynostosis Treatment & Management. En: Medscape (sede web). New York: Topol E; 2012 [en línea] [consultado el 20/10/2013]. Disponible en: http://emedicine.medscape.com/article/1175957-overview
- Scott JR, Isom CN, Gruss JS, Salemy S, Ellenbogen RG, Avellino A, et al. Sympton outcomes following cranial vault expansión for craniosynostosis in children older tan 2 years. Plast Reconstr Surg. 2009;123:289-97.
- Losee JE, Mason AC. Deformational plagiocephaly: diagnosis, prevention and treatment. Clin Plast Surg. 2005;32(1):53-64 [en línea] [consultado el 20/10//2013]. Disponible en: http://www.sciencedirect.com/science/article/pii/S0094129804000550
- Laughlin J, Luerssen TG, Dias MS; Committee on Practice and Ambulatory Medicine, Section on Nuerological Surgery. Prevention and mangement of positional skyll deformities in infants. Pediatrics. 2011;128:1236-41.
- Jiménez DF, Barone CM. Multiple-suture nonsyndromici craniosynostosis: early and effective management using endoscopic techniques. J Neurosurg Pediatr. 2010;5(3):1439-44 [en línea] [consultado el 20/10/2013]. Disponible en: http://thejns.org/doi/pdf/10.3171/2009.10.PEDS09216
- Shah MN, Kane AA, Peterson JD, Woo AS, Naidoo SD, Smith MD. Endoscopically assisted versus open repair of sagital craniosynostosis: the St. Louis Childrens Hospital experience. J Neurosurg Pediatr. 2011;8:165-70.