


Inmunodeficiencias primarias: aproximación diagnóstica
RESUMEN: PUNTOS CLAVE PARA EL PEDIATRA DE ATENCIÓN PRIMARIA
SUMMARY: KEY POINTS FOR PRIMARY CARE PEDIATRICIANS
PUNTOS CLAVE
- Las inmunodeficiencias primarias (IDP) son enfermedades raras, pero en general asociadas a una gran morbimortalidad, por lo que es fundamental un alto índice de sospecha para llegar a un diagnóstico precoz.
- Las manifestaciones clínicas más características son las infecciones de repetición, aunque no son las únicas.
- Las IDP más frecuentes son las que afectan a la inmunidad humoral, y suelen asociarse a infecciones del área otorrinolaringológica (ORL) y respiratorias de repetición a partir de los seis meses de vida.
- Las IDP combinadas, menos frecuentes, suelen aparecer en edades más tempranas y con formas clínicas más graves.
- El pediatra de Atención Primaria, mediante una anamnesis minuciosa, una exploración física completa y pruebas de laboratorio sencillas y accesibles, puede orientar un diagnóstico de IDP.
- En esos casos, ante la sospecha de una IDP es necesario remitir a una unidad especializada en Inmunología de forma precoz para completar el estudio diagnóstico e iniciar el tratamiento adecuado.
- El tratamiento debe individualizarse en función del diagnóstico, pero en todas las IDP es fundamental el manejo agresivo de las infecciones agudas y el tratamiento profiláctico (con Ig periódicas, antibioterapia, vacunación). El único tratamiento curativo en la actualidad es el trasplante de progenitores hematopoyéticos alogénico.
RESUMEN
Las IDP son un grupo de enfermedades causadas por defectos en los diferentes mecanismos implicados en la respuesta inmunitaria. Son enfermedades poco frecuentes, pero en general están asociadas a una gran morbimortalidad, sobre todo si se retrasa su diagnóstico. Este es difícil, y requiere en muchas ocasiones técnicas de laboratorio complejas y/o estudios genéticos pero, en muchos casos, una anamnesis minuciosa, una exploración física completa y pruebas de laboratorio sencillas accesibles desde Atención Primaria son suficientes para identificar los pacientes que precisan de un estudio inmunológico más completo.
INTRODUCCIÓN Y etioPATOGENIA
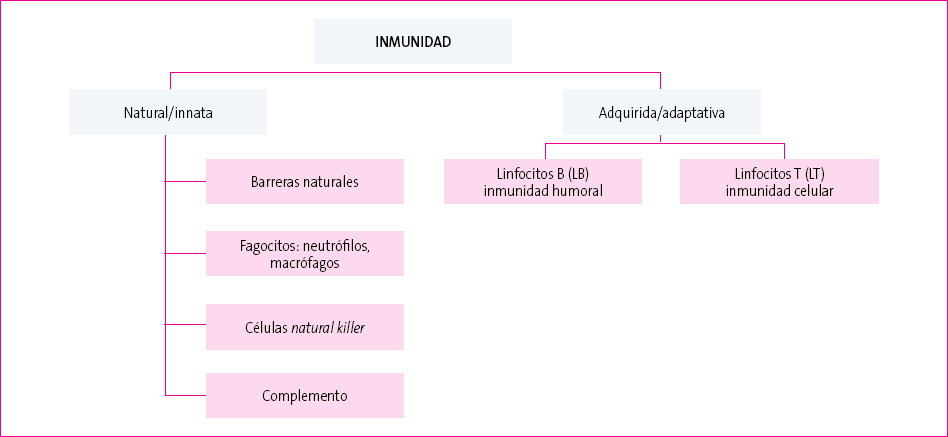
Las IDP son un grupo de enfermedades causadas por la alteración cuantitativa y/o funcional de los distintos mecanismos implicados en la respuesta inmunológica. Esta respuesta se puede dividir en natural o innata, y en adquirida o adaptativa (Figura 1).
Figura 1. Componentes del sistema inmunitario. Mostrar/ocultar
Son enfermedades poco frecuentes, con una incidencia estimada de 1/5000-10 000 nacimientos. El patrón de herencia es mayoritariamente autosómico recesivo (80-85% de los casos), aunque también se han descrito casos ligados al cromosoma X y de patrón autosómico dominante. La incidencia es mayor en determinadas etnias, en las que la consanguineidad es más prevalente. Existe un trastorno inmunitario mucho más frecuente, el déficit aislado de IgA, con una incidencia estimada 1/500-1000 nacidos, que en la mayoría de los casos cursa sin sintomatología.
La mayoría debutan en la infancia, pero algunas IDP se manifiestan en la edad adulta, como la inmunodeficiencia común variable (ICV), que se diagnostica fundamentalmente en la tercera o cuarta décadas de la vida.
Hasta la actualidad se han descrito alrededor de 200 déficits inmunitarios, que se clasifican en función de la estructura del sistema inmunitario afectado (Tabla 1).
Tabla 1. Clasificación de las inmunodeficiencias primarias. Mostrar/ocultar

MANIFESTACIONES CLÍNICAS
Los pacientes afectos de IDP presentan una gran variabilidad clínica, en función del defecto inmunitario presente. El cuadro clínico más frecuente son las infecciones de repetición y/o producidas por gérmenes poco habituales.
Como queda reflejado en la clasificación de las IDP (Tabla 1), más de la mitad se producen por trastornos de los linfocitos B. En estos casos las infecciones suelen aparecer entre los cuatro y los seis meses (no antes por la presencia de Ig trasplacentarias) y son mayoritariamente producidas por bacterias encapsuladas (S. pneumoniae, H. influenzae) en el tracto respiratorio (bronquitis, neumonía) y área ORL (otitis media aguda [OMA], sinusitis). Estas infecciones son, por otro lado, muy comunes en la edad pediátrica, lo que en ocasiones dificulta el diagnóstico de IDP.
En el caso de las IDP combinadas, la sintomatología suele ser más grave y precoz, como en el caso de la inmunodeficiencia combinada grave (SCID, por sus iniciales en inglés).
Otras manifestaciones clínicas que pueden aparecer son el retraso ponderal y/o la diarrea persistente, la presencia de citopenias no explicadas, fenómenos de autoinmunidad, lesiones cutáneas de difícil control y predisposición a tumores de estirpe linfoide, entre otras.
DIAGNÓSTICO
Dada la poca especificidad de algunas de las manifestaciones clínicas (OMA, bronquitis, dermatitis…), en muchas ocasiones el diagnóstico de las IDP supone un reto para el pediatra. Además, no siempre que existe una sospecha de IDP esta se confirma. Aproximadamente la mitad de los pacientes derivados a consultas especializadas en defectos inmunológicos por sospecha de IDP no presentan ningún trastorno inmunitario.
Una anamnesis exhaustiva, una exploración física completa y minuciosa y pruebas de laboratorio sencillas (hemograma y dosificación de inmunoglobulinas) constituyen los pilares fundamentales que permitirán establecer los pacientes con riesgo de presentar una IDP y que deben ser estudiados de forma más profunda.
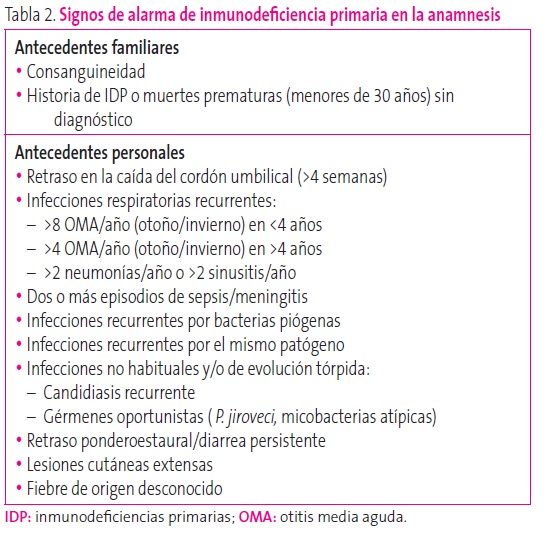
Signos de alarma en la anamnesis
La presencia de infecciones de repetición, con evolución tórpida de una infección en un niño y/o en alguno de sus familiares, debe hacernos pensar en una etiología inmunitaria.
En la Tabla 2 se resumen los datos en la anamnesis que puede ser sugestivos de la presencia de una IDP, según las recomendaciones de 2006 de la Sociedad Europea de Inmunodeficiencia (ESID).
Tabla 2. Signos de alarma de inmunodeficiencia primaria en la anamnesis. Mostrar/ocultar
Signos de alarma en la exploración física
En la mayoría de los casos no hay un fenotipo especial. En todos los pacientes es fundamental realizar una exploración minuciosa y completa, que incluya peso, talla y perímetro cefálico.
La presencia de signos en la piel y anejos nos puede aportar datos: albinismo(sugestivo de síndrome de Chediak-Higashi, síndrome de Griscelli), eccema de difícil control (presente en el síndrome de Wiskott-Aldrich, el síndrome de híper-IgE, inmunodisregulación, poliendocrinopatía y enteropatía ligado al cromosoma X [IPEX] o SCID tipo Ommen, entre otros). La presencia de granulomas de repetición orienta a un trastorno de los fagocitos. El hallazgo de vitíligo y/o alopecia sugiere un fenómeno de autoinmunidad asociado a IDP.
En el área ORL, los pacientes con IDP pueden presentar tanto hipertrofia como ausencia de tejido amigdalar. La presencia de úlceras orales, gingivitis de repetición y/o candidiasis recurrente orienta hacia la posibilidad de una IDP. Algunas malformaciones dentales se asocian típicamente a IDP (incisivos cónicos/displasia ectodérmica anhidrótica).
La presencia de hepatoesplenomagalia, adenopatías y lesiones del área perianal y periungueal deben alertar al pediatra sobre la posibilidad de una IDP que afecta los fagocitos.
Cuando un paciente presenta alguno de los signos de alarma anteriormente citados, sería necesario realizar estudios de laboratorio. Dado que no se pueden, ni se deben, realizar todos los estudios en un enfermo, la evaluación inicial analítica más adecuada consistiría en la realización de pruebas analíticas básicas, como un hemograma y niveles de inmunoglobulinas. Una adecuada interpretación de estas pruebas puede aportar mucha información sobre los trastornos primarios del sistema inmunitario.
Pruebas complementarias de primera línea
Hemograma
Siempre es necesario revisar todos los parámetros hematológicos y comprobar la existencia de hemogramas previos para determinar si el hallazgo es aislado y puntual o crónico (que sería más sugestivo de una IDP):
-
Serie blanca: valorar siempre las cifras en número absoluto y en función de la edad del paciente. Los parámetros a controlar en caso de una sospecha de IDP son los linfocitos y los neutrófilos:
- Linfocitos: la linfopenia mantenida (en al menos dos determinaciones analíticas) es el dato más orientativo de undéficit celular LTo combinado LT/LB. Es importante recordar que en los niños existe una linfocitosis fisiológica, por lo que se considera linfopenia cifras menores de 3000/μl en menores de dos años, por debajo de 2000/μl en niños de 3-5 añosy menores de 1500/μl en mayores decinco años.
- Neutrófilos: al igual que los linfocitos, es necesario valorar los resultados en número absoluto y en función de la edad y la raza. En menores de un año se considera neutropenia cifras menores de 1000/µl. En el resto de edades se define como neutropenia las cifras menores de 1500/µl. Las IDP que afectan a estas células predisponen a infecciones por bacterias y hongos. El hallazgo de neutropeniapuede orientara un déficit en fagocitos, pero un número normal no excluye la presencia de una IDP (neutropenia cíclica, enfermedad granulomatosa crónica).
- Serie roja:es necesarioexplorar todos los parámetros, hemoglobina, hematocrito, volumen corpuscular medio, hemoglobina corpuscular media, concentración de hemoglobina corpuscular media, amplitud de distribución eritrocitaria,en función de la edad. Si aparece anemia puede tener múltiples etiologías: carencial, trastornos crónicos, inmunitaria…
- Serie plaquetar:en las IDP pueden aparecer trombocitosis (habitualmente reactiva) o trombopenia. Si aparece esta última en el contexto de una IDP celular, puede ser de etiología inmunitaria.
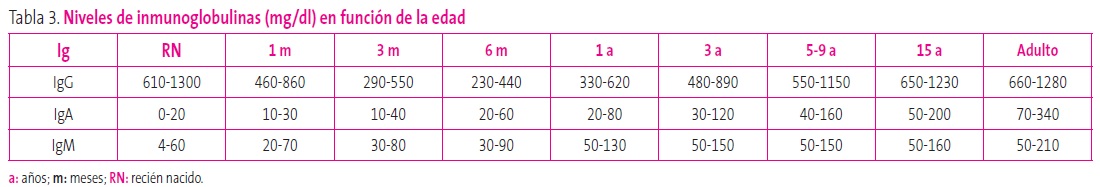
Niveles de inmunoglobulinas
Aportan datos sobre la inmunidad humoral (LB) o inmunidad combinada (LT/LB). Su interpretación es difícil antes de los 4-6 meses, debido al paso de inmunoglobulinas transplacentarias. Como en el caso del hemograma, siempre deben valorarse en función de la edad del paciente (Tabla 3).
Tabla 3. Niveles de inmunoglobulinas (mg/dl) en función de la edad. Mostrar/ocultar
En la mayoría de los casos, la combinación de los signos de alarma previamente citados y/o las alteraciones en alguna de estas pruebas de laboratorio nos permiten orientar las exploraciones de segunda línea que es necesario realizar para un diagnóstico definitivo, y que precisan de una valoración por un inmunólogo y de un laboratorio más especializado.
Pruebas complementarias de segunda línea
Se dividen en estudios cuantitativos, cualitativos y genéticos. Se ajustará la realización de unas u otras en función de la sospecha diagnóstica.
Estudios cuantitativos
- Fenotipaje linfocitario (poblaciones linfocitarias): mediante citometría de flujo se identifican los linfocitos en función de la expresión de determinados antígenos de superficie. De forma rutinaria, se identifican las siguientes subpoblaciones: CD3+CD4+ (linfocitos T colaboradores), CD3+CD8+ (linfocitos T citotóxicos/supresores), CD19+ (linfocitos B), CD16+CD56+ (células natural killer [NK]). En función de la sospecha clínica, se podrían realizar otros marcadores: CD25, CD45RO, CD45RA, CD40, CD40L... Su interpretación es difícil por su variabilidad a lo largo de la infancia, que hace necesario tener valores de referencia. Hay que tener en cuenta, además, que los valores absolutos de las subpoblaciones varían con la edad, aunque el porcentaje orientativo sería el siguiente: CD3 70%, CD4 40%, CD8 30%, CD19 20%, CD56 10%. Por otra parte, los valores están sometidos a múltiples variaciones, relacionadas con la presencia de infecciones, estrés, etc.
- Subclases IgG: deben realizarse preferiblemente en mayores de 18 meses, ya que antes de esta edad su interpretación es difícil y poco valorable. Es importante recordar que alrededor de un 20% de la población tiene niveles bajos desubclases de IgG con IgG normal, con lo que la alteración analítica no implica la presencia de una IDP. Habría que sospechar si asocia anamnesis o exploración de alarma y otras pruebas alteradas, como la respuesta a vacunasanómala. Dentro de los defectos de subclases de IgG, elmás frecuente es el déficit IgG2.
- Niveles IgE: elevado en algunas IDP, como el síndrome híper-IgE.
- Complemento: se debe sospechar IDP por déficit de complemento en pacientes con infecciones bacterianas de repetición (Neisseria, Haemophilus influenzae tipo b) con exploraciones de primera línea normales.
Estudios cualitativos
- Serologías vacunales: permiten constatar la capacidadd de producción de anticuerposespecíficos contra algunos gérmenes. Es necesario interpretarlas con prudencia en menores de seis meses por la presencia de Ig trasplacentarias. Se estudian dos tipos de anticuerpos: los antiproteicos, que aportan información sobre la capacidad de cooperación LT/LB,y los antipolisacáridos, que midenla capacidad de respuesta de los LB.
- Estudio funcional de linfocitos T: sirven para medir la capacidad de proliferación de los LT ante diferentes estímulos:sin inmunización previa (mitógenos) o con inmunización previa (antígenos).
- Estudio funcional de fagocitos: aportan datos sobre la capacidad de movimiento, adhesión y endocitosis de los polimorfonucleares (PMN). Son útiles en el estudio de trastornos cualitativos de PMN: enfermedadgranulomatosa crónica (test NBT, CF DHR123) y déficit de adhesión leucocitaria, entre otros.
Estudios genéticos
Se realizaran en los casos en los que esté descrita una mutación génica asociada a una IDP concreta.
Tratamiento
En relación al tratamiento de las IDP, hay que distinguir varios niveles:
- Tratamiento agresivo de las de infecciones agudas con antibioterapia precoz.
- Profilaxis de infecciones: en los trastornos de los fagocitos y en las IDP celulares/combinadas se emplean antibióticos como trimetropin-sulfametoxazol para evitar la infección por gérmenes oportunistas como el P. jiroveci. Respecto a las vacunas, es necesario individualizarlas en función del trastorno inmunitario que presente el paciente. Como las IDP representan un grupo muy heterogéneo respecto a la susceptibilidad a infecciones y la respuesta a inmunizaciones, la eficacia, seguridad y contraindicaciones de las diferentes vacunas dependerán del tipo y el grado de inmunosupresión de cada entidad. Existe un documento de consenso de la Sociedad Española de Infectología Pediátrica y el Comité Asesor de Vacunas de la Asociación Española de Pediatría para la vacunación en inmunodeprimidos, publicado en 2011, donde se especifican las recomendaciones vacunales en estos enfermos.
- Tratamiento sustitutivo: se emplea en los déficits de la inmunidad humoral, mediante la administración de inmunoglobulinas por vía intravenosa o subcutánea de forma periódica.
- Tratamiento etiológico: el único tratamiento curativo en la actualidad es el trasplante de progenitores hematopoyéticos alogénico, aunque existen datos sobre pacientes con determinadas IDP sometidos a terapia génica con resultados muy esperanzadores. En un futuro próximo se podrá obtener una reconstitución definitiva del sistema inmunitario mediante esta terapia.
BIBLIOGRAFÍA RECOMENDADA
- de Vries E; Clinical Working Party of the European Society forImmunodeficiencies (ESID). Patient-centred screening for primary immunodeficiency: a multi-stage diagnostic protocol designed for non-immunologists. Clin Exp Immunol. 2006;145:204-14.
- García Martínez JM, Santos-Díez L, Dopazo L. Diagnóstico de las inmunodeficiencias primarias. Protoc diagn ter pediatr. 2013;1:81-92. En aeped.es [en línea]. Disponible en: http://www.aeped.es/sites/default/files/documentos/7-inmunodeficiencias_primarias_0.pdf
- Mellado Peña MJ, Moreno Pérez D, Ruiz Contreras J, Hernández-Sanpelayo Matos T, Navarro Gómez ML; grupo de colaboradores del Documento de Consenso SEIP-CAV de la AEP. Documento de consenso de la Sociedad Española de Infectología Pediátrica y el Comité Asesor de Vacunas de la Asociación Española de Pediatría para la vacunación en inmunodeprimidos. An Pediatr (Barc). 2011;75:413.e1-413.e22.
- NotarangeloLD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125(Suppl 2):S182-94.
- Oliveira JB, Fleisher TA. Laboratory evaluation of primary immunodeficiencies. J Allergy Clin Immunol. 2010;125(Suppl 2):S297-305.
- Picard C. Comment explorer un déficit immunitaire héréditaire? La Revue du Practicien. 2007;57.
- Stiehm ER. Approach to the child with recurrent infections. En: UpToDate [en línea] [actualizado el 14/09/2012]. Disponible en: http://www.uptodate.com/contents/approach-to-the-child-with-recurrent-infections
Cómo citar este artículo
Artículos relacionados
 Infecciones recurrentes y sospecha de inmunodeficiencias
Infecciones recurrentes y sospecha de inmunodeficiencias
Albañil Ballesteros MR. Infecciones recurrentes y sospecha de inmunodeficiencias. Form Act Pediatr Aten Prim. 2011;4;25-30