



Infecciones recurrentes y sospecha de inmunodeficiencias
RESUMEN: PUNTOS CLAVE PARA EL PEDIATRA DE ATENCIÓN PRIMARIA
SUMMARY: KEY POINTS FOR PRIMARY CARE PEDIATRICIANS
PUNTOS CLAVE
- Las infecciones frecuentes pueden ser una situación normal en algunas épocas de la infancia.
- La mayoría de los niños que las presentan son sanos.
- Las infecciones de repetición pueden expresar otra situación patológica distinta de una inmunodeficiencia (ID).
- Es preciso conocer los datos de alarma que hagan sospechar una ID.
- La importancia del diagnóstico precoz se basa en la prevención de daños crónicos y en que alguna entidad, como la inmunodeficiencia severa combinada, se trata de una emergencia médica.
- Es preciso conocer otras manifestaciones posibles de ID: enfermedades autoinmunes y malignas.
- La historia familiar es clave.
- Una vez orientado el diagnóstico de ID o con alta sospecha del mismo, es preciso la consulta con el experto.
INFECCIONES RECURRENTES
La patología infecciosa es la causa más frecuente de consulta en Pediatría de Atención Primaria y Servicios de Urgencia. En algunas épocas de la vida, un número importante de niños presenta un agrupamiento en el tiempo de estos procesos y, si bien la mayoría de ellos son sanos, es preciso distinguir quiénes son susceptibles de ser estudiados para descartar una ID. El objetivo de este trabajo no es realizar una enumeración exhaustiva de todas las ID ni realizar una presentación de cada una de ellas, sino establecer unas orientaciones prácticas acerca de qué es lo esperable en niños normales, en qué circunstancias cabe sospechar una ID y cómo debe orientarse su estudio, este apartado será objeto de un segundo artículo.
Los niños que presentan infecciones frecuentes pueden incluirse en uno de los grupos que figuran en la tabla 1 y se detallan a continuación1.
Tabla 1. Mostrar/ocultar

Niños normales
Es el grupo más frecuente. Son niños que presentan infecciones recurrentes sin causa orgánica, funcional, inmunitaria o genética conocida2. El motivo es la frecuente exposición a microorganismos infecciosos3 a los que se enfrentan por primera vez.
En sus primeros años de vida, los niños normales pueden presentar de cuatro a ocho episodios infecciosos al año, y hasta 10-12 si están escolarizados, asisten a guardería, tienen hermanos mayores o están expuestos al humo del tabaco. Lo habitual es que no tengan más de una neumonía y dos episodios de otitis media aguda (OMA) anuales, en los primeros tres años de vida, pero pueden considerarse normales 6-8 infecciones respiratorias de vías altas (IRVA) cada año, hasta seis episodios anuales de OMA y dos de gastroenteritis aguda (GEA) en los 2-3 primeros años4. La duración media de cada proceso es de ocho días, pero puede llegar a las dos semanas y, dado que se agrupan estacionalmente, el niño puede presentar síntomas durante largo tiempo, dando la impresión de que siempre está enfermo. La mayoría de los procesos son de etiología viral, afectan al tracto respiratorio y responden en tiempo y forma habitual a los tratamientos adecuados, los niños tienen un crecimiento y un desarrollo normales, entre episodios están bien y la exploración y las pruebas complementarias son normales.
Niños atópicos
Son niños que presentan el espectro alergia-atopia-sibilancias recurrentes-asma, son el segundo grupo en frecuencia. Aunque se trata de cuadros de etiología no infecciosa, rinitis, cuadros con sibilancias y tos, pueden confundirse con infecciones porque presentan algunos síntomas comunes y pueden exacerbarse y desencadenarse por procesos infecciosos. Según algunos autores, podrían facilitar la existencia de sinusitis, OMA, etc., con tendencia a repetir el mismo proceso en el mismo niño. Deben responder, según la clínica, a broncodilatadores o antihistamínicos, y no a antibióticos. En general, son niños con crecimiento y desarrollo normales y con algún rasgo fenotípico propio1.
Niños crónicamente enfermos
Estos niños presentan enfermedades crónicas no inmunitarias u otras situaciones que favorecen la infección. Incluyen situaciones muy variadas y diversas entre sí, desde defectos anatómicos localizados, fístulas, problemas obstructivos, fallos de barrera, ausencia de aclaramiento de secreciones, existencia de cuerpo extraño accidental o catéteres implantados, hasta enfermedades crónicas como fibrosis quística (FQ), cardiopatías con hiperaflujo, encefalopatías, miopatías, diabetes o síndrome nefrótico, entre otras. Algunas, como la drepanocitosis o el síndrome nefrótico, presentan una susceptibilidad especial a gérmenes concretos como el neumococo.
Estos niños pueden presentar afectación del crecimiento y el desarrollo y hallazgos físicos en consonancia con la causa subyacente, pero son un grupo muy heterogéneo y las manifestaciones que presentan, muy variadas1.
Niños inmunodeficientes
Representan un pequeño porcentaje, y en ellos las infecciones se deben a una alteración cuantitativa y/o funcional de uno o varios mecanismos implicados en la respuesta inmunitaria2.
Niños que presentan síndromes que cursan con fiebre periódica o recurrente
Dado que el síntoma capital de muchas entidades aquí agrupadas es la fiebre, deben incluirse en el diagnóstico diferencial de las infecciones recurrentes. Se agrupan aquí muchas entidades muy poco frecuentes cuyo diagnóstico es difícil de realizar.
De modo arbitrario, se define fiebre recurrente o periódica como tres o más episodios de fiebre en seis meses y con intervalo libre entre los episodios febriles5. Puede distinguirse entre intervalo típicamente regular o irregular. Con intervalo regular: síndrome PFAPA (Periódica Fiebre Aftosa Faringitis Adenopatía Cervical), que cursa con leucocitosis y la neutropenia cíclica, cuadro similar pero con neutropenia. Con intervalo ocasionalmente regular, pero más típicamente irregular, algunos síndromes autoinflamatorios: fiebre mediterránea familiar y síndrome Hiper-IgD e infecciones por virus de Epstein-Barr. Entre las causas de fiebre recurrente a intervalos irregulares hay causas víricas, bacterianas y parasitarias, neoplasias, fiebre por fármacos, facticia y por alteraciones del sistema nervioso central, así como enfermedades inflamatorias y síndromes hereditarios autoinflamatorios. La existencia de tres o más episodios de fiebre sucesivos, sin signos de infección viral, debe sugerir un diagnóstico alternativo. Datos clínicos que pueden presentarse en estos síndromes son: úlceras orales o genitales, artralgias y artritis, adenopatía cervical, dolor abdominal y lesiones de la piel, exantemas morbiliformes coincidiendo o no con la fiebre, máculo-pápulas, petequias, eritema erisipela like, eritema nodoso, uveítis y conjuntivitis dolorosa, sordera neurosensorial, pleuritis, pericarditis, esplenomegalia, mialgias y aparición de la sintomatología tras inmunización o exposición al frío5,6. En la última clasificación de las ID según la International Union of Immunological Societies (IUIS)7, estos síndromes aparecen clasificados como un grupo de ID. En la tabla 2 figura una clasificación de causas de fiebre recurrente.
Tabla 2. Mostrar/ocultar

SOSPECHA DE INMUNODEFICIENCIAS
Las ID son entidades en las que hay afectación cuantitativa o cualitativa de cualquier componente del sistema inmunitario: inmunidad adaptativa (células B o inmunidad humoral o respuesta de anticuerpos y células T o sistema celular) e inmunidad innata (del sistema fagocítico y del complemento, entre otros). Según el lugar de la diferenciación y maduración en que se produzca la alteración, desde célula madre pluripotencial a receptores, estimuladores o enzimas, se producirán distintos defectos7. Los pacientes que presentan ID pueden padecer infecciones por gérmenes atípicos, oportunistas, o por gérmenes habituales pero persistentes; estas infecciones serán recurrentes, más severas, con mala respuesta al tratamiento adecuado, a veces en localizaciones características y, en todo caso, con evolución inhabitual. Y tienen susceptibilidad elevada ante enfermedades malignas y autoinmunitarias1.
Hay hasta 200 entidades descritas y su número aumenta, con un rango de frecuencias entre 1/10 000 y 1/200 000, y de 1/2000 a 1/10 000 de todas las ID consideradas conjuntamente, excluyendo el déficit de IgA, que es 1/300-700 en caucásicos8.
La mayoría de las ID Primarias son hereditarias, suelen manifestarse en el primer año de vida y afectan más frecuentemente a las células B.
Las ID secundarias son más frecuentes que las primarias. Afectan más a las células T. En ellas, la alteración de la función inmunitaria se produce por medicaciones inmunosupresoras utilizadas en trasplantes, tratamiento de enfermedades autoinmunes o malignas, malnutrición, por infecciones por agentes como el virus de la inmunodeficiencia humana (VIH), síndromes pierde-proteínas, y enfermedades neoplásicas o de otro tipo como diabetes mellitus. Infecciones perinatales por citomegalovirus y otros virus herpes pueden causar inmunodepresión transitoria o permanente1.
De las alteraciones primarias, más del 50% afecta a la inmunidad humoral (células B), el 8% a la inmunidad humoral y celular (células T), el 18% corresponde a defectos bien definidos, el 11% a defectos de los fagocitos, el 2,5% a defectos genéticos de la regulación, el 2,5% a defectos del complemento, mientras que los síndromes autoinflamatorios y los defectos de la inmunidad innata corresponden a un 0,4% cada uno9. La entidad más frecuente es el déficit de IgA, que es asintómatico en la mayoría de los casos, pero puede cursar con infecciones respiratorias y gastrointestinales recurrentes, asociar enfermedades autoinmunitarias y presentar reacciones anafilácticas frente a hemoderivados. En algún caso puede progresar a inmunodeficiencia variable común (IDVC)10. Aunque clásicamente las ID se clasificaban en cinco grupos según afectaran a la inmunidad celular, humoral, de forma combinada a ambas, al sistema fagocítico y al complemento, la última clasificación de la IUIS considera ocho grupos. En la tabla 3 figura esta clasificación resumida, con algunos de los cuadros incluidos en cada grupo.
Tabla 3. Mostrar/ocultar

Para un conocimiento más completo del tema, se recomienda consultar este artículo que incluye también datos clínicos, defectos funcionales, herencia y frecuencia7.
Datos de sospecha de ID
Infecciones recurrentes: es difícil señalar como límite exacto un número total de infecciones al año por encima del cual la situación se considere patológica. Algunos autores establecen 11-12 infecciones respiratorias agudas y 14 episodios de OMA y/o gastroenteritis11. La mayoría dan más valor al tipo de infecciones y a las características de las mismas: órganos y sistemas afectados, localización, gravedad y germen responsable.
En cuanto a la fecha de inicio de las infecciones, hay ID que ya en época neonatal comienzan a producir clínica o a mostrar signos que pueden hacer sospechar su existencia, como neutropenia congénita, inmunodeficiencia severa combinada (IDSC), síndrome de Di George, defectos en TLR3 –con encefalitis herpética neonatal–, déficit de IR-1 receptor asociado a cinasa 4 (IRAK 4), defectos de adhesión leucocitaria (LAD) con úlceras perianales y retraso en la caída del cordón, enfermedad granulomatosa crónica (EGC), síndrome de Omenn con eritrodermia, linfadenopatía y esplenomegalia, y pancitopenia en la disgenesia reticular. Hay otras que pueden iniciarse más tardíamente, incluso en jóvenes adultos, como inmunodeficiencia variable común (IDVC) y algunas formas de EGC, agammaglobulinemia de Bruton y alteraciones del complemento. En las ID de predominio humoral, la presencia de anticuerpos de origen materno pueden proteger al niño en los primeros meses de vida y retrasar la aparición de síntomas1,4,12. La enfermedad linfoproliferativa ligada al cromosoma X puede tener un inicio variable13. En las inmunodeficiencias secundarias, la clínica aparece después de instaurarse la causa que las origina.
Una historia familiar de ID conocida o de muertes inexplicadas o precoces también debe orientar a la sospecha.
Tipos de infección: no todas las infecciones tienen la misma importancia a la hora de sospechar que sean indicativas de un estado de inmunodeficiencia. Las rinitis, faringoamigdalitis, laringitis e infecciones del tracto urinario (ITU) no lo son. Impétigos y forúnculos pueden tener relación con falta de higiene, aunque algunas ID cursan con cuadros de eccema severo y posible sobreinfección, y otras con abscesos cutáneos. Bronquitis, conjuntivitis y OMA habitualmente no lo son, pero también pueden verse en cuadros de ID, sobre todo humoral. Puede existir, además, otra causa que motive su recurrencia: cuerpo extraño en rinitis unilaterales, alteraciones anatómicas en ITU, asma en bronquitis, FQ entre otras.
Sí deben alertar las sinusitis y neumonías. En estas últimas, si son en la misma localización, puede existir otro motivo facilitador como cuerpo extraño, adenopatía, anomalía vascular, secuestro o quiste. En distinta localización puede tratarse de asma, FQ, reflujo gastroesofágico o enfermedad por cilio inmóvil. A veces, las ID cursan con síntomas crónicos que pueden producir un deterioro pulmonar progresivo. Otras infecciones que deben alertar sobre existencia de ID son las diarreas crónicas, y más si asocian alteración del crecimiento, estomatitis persistentes, candidiasis oral en niños mayores en ausencia de tratamiento antibiótico o con corticoides inhalados, e infecciones mucocutáneas graves y frecuentes, ulceración perianal, particularmente neonatal, abscesos de piel, ganglios y órganos internos, sepsis y meningitis, aunque también pueden deberse a una alteración anatómica que produzca comunicación1,4. Las úlceras orales recurrentes pueden verse en enfermedades autoinmunitarias y síndromes de fiebre periódica y neutropenia13.
Gérmenes. Según el tipo de defecto inmunitario pueden verse implicados determinados gérmenes como causantes de patología, aunque una respuesta humoral (de células B) adecuada precisa integridad de la inmunidad celular (de células T) y, por tanto, es posible ver conjuntamente manifestaciones por defectos de ambas líneas. En la tabla 4 figuran el tipo de gérmenes y las patologías más frecuente que se observan con cada uno de los grupos clásicos de ID; además, algunos patógenos o infecciones se asocian de forma característica con entidades concretas. En el documento de la IUIS7 y las revisiones de Slatter12 y Ballow13, puede consultarse una más amplia relación de entidades concretas con la patología típicamente asociada a cada una de ellas.
En la tabla 5 se recogen datos concretos descritos como signos de alarma en la literatura.
Tabla 4. Mostrar/ocultar
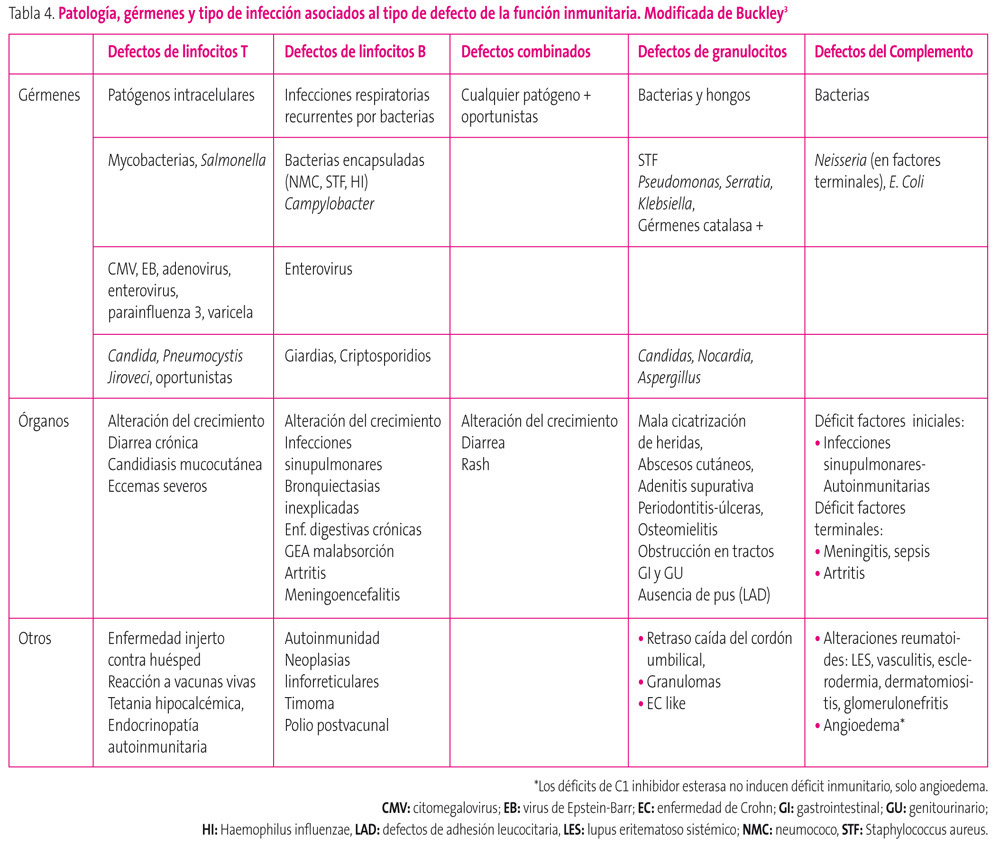
Tabla 5. Mostrar/ocultar

En general, es raro que se trate de ID si no hay infecciones profundas documentadas, el niño presenta un crecimiento y desarrollo adecuado, permanece sano entre episodios y no hay historia familiar de ID conocida o datos de sospecha de la misma11.
BIBLIOGRAFÍA
- Stiehm R. Approach to the child with recurrent infections. www.Uptodate.com. Última revisión mayo 2010 [Consultado el 17-10-2010].
- Martín Nalda A. Infecciones de repetición: cuántas son demasiadas. Signos de alarma de inmunodeficiencias primarias en pediatría. En: Libro de ponencias del 58 Congreso de la Asociación Española de Pediatría. Zaragoza; 2009. p. 396-8.
- Buckley R. Evaluación del sistema inmunitario. En: Kliegman RM, Behrman E, Jenson HB, Stanton BF (eds.). Nelson tratado de pediatría, 18.ª ed. Elsevier; 2009. p. 867-73.
- Ruiz Contreras J. El niño con infecciones frecuentes. En: AEPap ed. Curso de Actualización Pediatría 2010. Madrid: Exlibris Ediciones; 2010. p. 15-22.
- Chandy J, Gilsdorf J. Recurrent fever in children. Pediat Infect Dis J. 2002;21:1071-80.
- Andreu Alapont E, Lacruz Pérez L, López Montesinos B. ¿Este niño tiene reuma? En: AEPap ed. Curso de Actualización Pediatría 2010. Madrid: Exlibris Ediciones; 2010. p. 245-58.
- International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies. Notarangelo L, Fischer A, Geha RS, Casanova JL, Chapel H, MD, Conley ME et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161-78.
- Bonilla FA, Bernstein I, Khan DA, Ballas ZK, Chinen J, Frank MM et al. Practice parameter for diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94:S1-64.
- Rezaei N, Bonilla F, Sullivan K, de Vries E, Orange J. An introduction to primary Immunodeficiency Diseases. En: Rezai N, Aghamohammadi A. Primary Immunodeficiency Diseases. Definition, diagnosis and management. Berlin: Springer-Verlag; 2008. p. 1-38.
- Hostoffer R. Selective IgA deficiency: Clinical manifestations, pathophysiology, and diagnosis. Uptodate. Última version mayo 2010 [consultado el 21-10-2010].
- Gurbindo MD. Infecciones recurrentes en el niño. Sospecha de inmunodeficiencia. En: Manrique I, Saavedra J, Gómez JA, Álvarez G (eds.). Guía de tratamiento de las Enfermedades Infecciosas en Pediatría, 3.ª edición. Madrid: 2010. p. 851-7
- Slatter MA, Gennery AR. Clinical Immunology Review Series: An approach to the patient with recurrent infections in chilhood. Clinical and Experimental Immunology. 2008;152:389-96.
- Ballow M. Approach to the Patient With Recurrent Infections. Clin Rev Allerg Immunol. 2008;34:129-40.
BIBLIOGRAFÍA RECOMENDADA
- Stiehm R. Approach to the child with recurrent infections. www.Uptodate.com. Última revisión mayo 2010 [consultado el 17-10-2010]. Aporta una interesante visión general del tema.
- International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies: Notarangelo L, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161-78. Documento actualizado con la clasificación exhaustiva de las inmunodeficiencias, así como con datos clínicos, analíticos, tipo de herencia y defecto molecular si se conoce.
NOTA
Con fecha posterior a la publicación del artículo, ha sido editado en Anales de Pediatría un documento de Consenso de la Sociedad de Infectología Pediátrica y la Sociedad de Reumatología Pediátrica (ambas de la AEP) sobre diagnóstico diferencial y el abordaje de la fiebre recurrente, que la autora considera puede ser de interés para el lector.
La referencia exacta del mismo es:
Calvo Rey C.; Soler-Palacín, P.; Merino Muñoz, R.; Saavedra Lozano, J.; Antón López, J.; Aróstegui, J.I.; Blázquez Gamero, D.; Martín-Nalda, A.; Juan, M.; Méndez, M.; Piñeiro Perez, R.; Calvo, I. Asociación Española de Pediatría. Documento de Consenso de la Sociedad de Infectología Pediátrica y la Sociedad de Reumatología Pediátrica sobre el diagnóstico diferencial y el abordaje terapéutico de la fiebre recurrente. An Pediatr (Barc). 2011;74(3):194.e1-194.e16.
Cómo citar este artículo
Artículos relacionados
 Inmunodeficiencias primarias: aproximación diagnóstica
Inmunodeficiencias primarias: aproximación diagnóstica
Rodríguez-Vigil Iturrate C. Inmunodeficiencias primarias: aproximación diagnóstica. Form Act Pediatr Aten Prim. 2014;7;75-80- Orientación al diagnóstico y tratamiento de las inmunodeficiencias
Albañil Ballesteros MR. Orientación al diagnóstico y tratamiento de las inmunodeficiencias. Form Act Pediatr Aten Prim. 2011;4;77-81