

Diagnóstico diferencial de la ictericia en el lactante y niño mayor
2 Pediatra. CS Actur Norte. Zaragoza. (España).
3 MIR-Pediatría. Hospital Universitario Miguel Servet. Zaragoza. (España).
PUNTOS CLAVE
- En todo niño con ictericia de más de 2 semanas de vida se deben medir los niveles de bilirrubina total y directa.
- A la hora del diagnóstico, es fundamental distinguir entre la hiperbilirrubinemia indirecta/no conjugada, hiperbilirrubinemia directa/conjugada. La anamnesis, el examen físico y las pruebas de laboratorio iniciales junto con esta clasificación se utilizan para orientar la etiología.
- La causa más frecuente de ictericia es el síndrome de Gilbert, patología benigna con una prevalencia del 3-7% y predominio masculino1.
- Sin embargo, la ictericia puede ser la primera manifestación clínica de una hepatopatía crónica no diagnosticada o la presentación de un fallo hepático agudo, por lo que es fundamental una correcta evaluación que incluya anamnesis exhaustiva con exploración física completa y las pruebas complementarias pertinentes.
- La atresia de vías biliares es la causa más frecuente de colestasis en el recién nacido o lactante sin patología neonatal. Por tanto, se debe descartar dicha etiología en todo lactante con ictericia prolongada (más de 15 días) mediante la medición de bilirrubina directa, ya que el retraso diagnóstico tiene implicaciones pronósticas muy graves2.
- El principal abordaje desde Urgencias se basa en la detección de una situación de potencial gravedad y la instauración del tratamiento de soporte en función de la etiología.
RESUMEN
La ictericia es la pigmentación amarillenta de piel y mucosas, siendo clínicamente evidente cuando los niveles de bilirrubina total sobrepasan los 2 mg/dl en lactantes y niños mayores, siendo poco frecuente fuera del periodo neonatal. Por ello, es importante una anamnesis exhaustiva y exploración física minuciosa que orienten la causa de esta. En función de la fracción de bilirrubina que se encuentre elevada se realizará el diagnóstico diferencial. El manejo terapéutico se realiza de acuerdo a la etiología, requiriendo en la mayoría de los casos el ingreso hospitalario para completar estudio.
INTRODUCCIÓN
La ictericia es un signo clínico caracterizado por la pigmentación amarillenta de piel y mucosas consecuencia del depósito de bilirrubina en dichos tejidos, siendo clínicamente evidente cuando los niveles de bilirrubina total sobrepasan los 2 mg/dl en lactantes y niños mayores. La concentración normal de bilirrubina sérica total en niños es menor a 1 mg/dl (bilirrubina directa <0,4 mg/dl).
Se trata de un signo clínico frecuente en el periodo neonatal, considerándose patológico fuera del mismo.
La ictericia fuera del periodo neonatal es causa poco frecuente de consulta en el servicio de atención primaria. Las formas más frecuentes son debidas a hiperbilirrubinemia indirecta producida por hemólisis y el síndrome de Gilbert. Por el contrario, la hiperbilirrubinemia no conjugada abarca etiología diversa de mayor relevancia y generalmente asociadas a mayor morbimortalidad.
FISIOPATOLOGÍA
La bilirrubina (Bb) es un pigmento biliar producto de la degradación del grupo hemo (80%) presente en la hemoglobina, la mioglobina y los citocromos, entre otros. Se distinguen dos tipos3.
La bilirrubina indirecta o no conjugada es aquella que circula en sangre unida a la albúmina y una pequeña fracción permanece libre en plasma. La unión con la albúmina mantiene la bilirrubina en el espacio vascular, lo que evita el depósito en los tejidos extrahepáticos, incluidos los tejidos sensibles, como el cerebro, así como la filtración glomerular al ser insoluble. La bilirrubina libre puede causar toxicidad cerebral cuando la concentración molar de la misma excede la de la albúmina3.
La Bb directa o conjugada se forma en los sinusoides hepáticos, el complejo albúmina-bilirrubina se disocia y el hepatocito absorbe la bilirrubina que se conjuga con el ácido glucurónico gracias a la enzima UGT para excretarse a los canalículos biliares, formando parte de la bilis. Las bacterias intestinales juegan un papel importante en la transformación a urobilinógeno y estercobilinógeno, modo en el cual se elimina en las heces. Parte de la bilirrubina vuelve al hígado mediante recirculación enterohepática3.
ETIOPATOGENIA
La clasificación etiológica de la ictericia se divide en función de la fracción de bilirrubina que se encuentra elevada.
La hiperbilirrubinemia indirecta/no conjugada: (aumento de Bb total, con Bb directa <20% del total) puede ser secundaria a1,4:
-
Sobreproducción de bilirrubina debida a extravasación, hemolisis extravascular o intravascular y eritropoyesis anormal.
- En la mayoría de la población, las anemias hemolíticas congénitas corpusculares son la causa más frecuente de hemólisis intravascular (hemoglobinopatías, talasemias, defectos de la membrana eritrocitaria y enzimopatías). Las anemias hemolíticas adquiridas de origen autoinmune, microangiopatías o debidas a fármacos son causas menos frecuentes.
- En la diseritropoyesis existe una incorporación defectuosa de hemoglobina a los glóbulos rojos, lo que conduce a la degradación de una gran fracción de la hemoglobina hemo no incorporada, como ocurre, por ejemplo, en anemias megaloblásticas y sideroblásticas, porfiria eritropoyética, eritroleucemia y envenenamiento por plomo, entre otras.
- Disminución de la captación hepática. La administración de diferentes medicamentos como la rifampicina, probenecid, la disminución del flujo sanguíneo hepático ocasionado en la insuficiencia cardiaca o en las derivaciones portosistémicas pueden alterar la captación hepática.
- Defecto de la conjugación, los trastornos hereditarios como el síndrome de Gilbert y el síndrome de Crigler-Najjar tipo I-II causan disminución o ausencia de la actividad de la enzima uridina difosfoglucuronato glucuroniltransferasa (UGT). Enfermedades adquiridas como el hipotiroidismo, hepatitis crónica o cirrosis avanzada pueden ocasionar alteraciones en la conjugación.
La hiperbilirrubinemia directa/conjugada o mixta (>1 mg/dl si Bb total <5 mg/dl o >20% si Bb total >5 mg/dl) puede ser secundaria a daño hepatocelular, alteración de la excreción canalicular, afectación de la vía biliar intra o extrahepática o a un defecto de la redistribución enterohepática1.
- Alteración hepatocelular. Las hepatitis víricas (VHA, VHB, VHC, CMV, VEB, TORCH, herpesvirus), bacterianas (E. coli) u otras, así como fármacos, tóxicos, drogas o enfermedades metabólicas tales como enfermedad de Wilson, galactosemia, tirosinemia, fibrosis quística, déficit de alfa 1 antitripsina, pueden causar daño hepático y, por consiguiente, aumento de niveles de bilirrubina.
- Afectación de la vía biliar. Atresia vías biliares, quiste del colédoco, colelitiasis, coledocolitiasis, colecistitis, colangitis, tóxicos.
- Defecto del transporte canalicular de aniones orgánicos, donde destaca el síndrome de Dubin-Jhonson.
- Defecto de la recaptación sinusoidal de bilirrubina conjugada, siendo el síndrome de Rotor causa infrecuente de ictericia.
DIAGNÓSTICO
El abordaje diagnóstico del paciente con ictericia comienza con una anamnesis exhaustiva, un examen físico completo y estudios de laboratorio iniciales.
La anamnesis dirigida debe recoger los siguientes datos5:
- Edad, sexo, raza, etnia.
- Tiempo de evolución y desencadenante de la ictericia. Coloración de la orina (coluria) y de las heces (acolia).
- Sintomatología asociada: fiebre, vómitos, dolor abdominal, deposiciones líquidas, astenia, prurito, hemorragias, anorexia, irritabilidad, alteraciones del comportamiento.
- Antecedentes personales: retraso en eliminación de meconio, íleo meconial, tipo de lactancia, ganancia ponderoestatural, desarrollo psicomotor, estado vacunal.
- Antecedentes familiares: consanguinidad, trastornos hereditarios (incluidos trastornos hemolíticos y enfermedades hepáticas), ictericia, anemia, hepatopatía, litiasis biliar, esplenectomía, enfermedades autoinmunes, síndrome de Gilbert.
- Consumo de tóxicos: fármacos, alcohol, drogas, productos de herbolario.
- Antecedentes dietéticos: alimentación con fructosa, lactosa, consumo de caroteno, ingesta de habas.
- Infecciones, viajes recientes a áreas endémicas de hepatitis, contacto con animales.
- Valorar los rasgos fenotípicos de los pacientes, debido a su alteración en algunos cuadros sindrómicos (Alagille, Zellgewer, hiopituitarismo).
En la exploración física debe constar5:
- Triángulo de evaluación pediátrica.
- Peso y talla.
- Constantes vitales (FC, FR, TA, SatO2, T).
- Estado general. Estado de hidratación (mucosas húmedas, llanto con lágrima, signo del pliegue...).
-
Piel y esclerótica: ictericia (zonas de Kramer), palidez, petequias, arañas vasculares, cefalohematomas, hematomas.
-
Se evaluará al paciente desnudo, preferentemente bajo luz natural en una habitación con iluminación adecuada. La afectación cutánea sigue una progresión cefalocaudal, por lo que algunos autores consideran que se pueden estimar de forma aproximada los niveles séricos de bilirrubina en función del área corporal afectada (Tabla 1).
Tabla 1. Zonas Kramer y valor sérico de bilirrubina 5. Mostrar/ocultar
- En pacientes con piel oscura, las palmas y plantas de los pies están menos pigmentadas y pueden ser más fáciles de evaluar en busca de ictericia o palidez. En pacientes con hiperbilirrubinemia leve, la ictericia puede ser sutil y objetivarse únicamente en la esclera.
- Debe hacerse diagnóstico diferencial con la carotenemia. En esta, las escleróticas no se decoloran y la coloración cutánea suele ser anaranjada. Además, no existe elevación sérica de bilirrubina.
-
Se evaluará al paciente desnudo, preferentemente bajo luz natural en una habitación con iluminación adecuada. La afectación cutánea sigue una progresión cefalocaudal, por lo que algunos autores consideran que se pueden estimar de forma aproximada los niveles séricos de bilirrubina en función del área corporal afectada (Tabla 1).
- Exploración por aparatos; neurológico (Glasgow, tono, comportamiento, temblor), ocular (anillos Kayser-Fleicher), exudado faringoamigdalar y linfadenopatías (mononucleosis infecciosa), estigmas hepáticos (arañas vasculares, eritema palmar, circulación colateral), cardiaco (soplos), abdominal (hepatoesplenomegalia, ascitis, vascularización colateral, signo de Murphy).
PRUEBAS COMPLEMENTARIAS
Las pruebas complementarias ayudan a establecer el diagnóstico etiológico, por lo que en todos los niños que presenten ictericia fuera del periodo neonatal se deberá completar el estudio con analítica sanguínea que incluya: hemograma, bioquímica (bilirrubina total y fraccionada, función hepática (GOT, GPT, FA, GGT), LDH, albúmina, proteínas totales, iones, colesterol, función renal) y coagulación.
- El aumento de FA, GGT y bilirrubina directa orientan a alteración de la vía biliar.
- La elevación de enzimas hepáticas (GOT/GPT) indican lesión hepatocelular como responsable de la ictericia.
- El tiempo de protrombina y albúmina evalúan la función hepática, afectándose en estadios avanzados de hepatopatía. Si se normaliza el tiempo de protrombina (alargado) tras la administración de vitamina K, sugiere colestasis.
La necesidad de añadir pruebas complementarias dependerá de la orientación diagnóstica del caso.
- Reactantes de fase aguda: en caso de sospecha de patología infecciosa.
- Serología: ante antecedentes y clínica sugestiva de hepatitis, añadir serologías de virus hepatotropos, VHA (IgM anti VHA), VHB (AgHbs, anti HBc IgM), VHC (ARN de VHC), VEB, CMV, mycoplasma.
- Estudio de hemólisis: ante hiperbilirrubinemia indirecta y anemia, completar estudio de parámetros de hemólisis que incluya: LDH, haptoglobina, reticulocitos, grupo sanguíneo, RH, test de Coombs directo, extensión de sangre periférica y básico de orina (hemoglobinuria). Si existen signos clínicos de anemia (taquicardia, palidez…) solicitar pruebas cruzadas.
- Tira de orina: para determinar hiperbilirrubinemia (conjugada) o en caso de sospecha de infección urinaria.
- Cultivos: si fiebre o síntomas/signos y exploración física compatible con sepsis o infección urinaria, extraer frotis nasofaríngeo, hemocultivo y urocultivo.
- Tóxicos en sangre u orina: si se sospecha como agente etiológico.
- Gota gruesa: paciente con fiebre, hiperbilirrubinemia indirecta y antecedente de viaje a zona endémica de malaria.
- Ecografía abdominal: ante aumento de hiperbilirrubinemia directa para descartar patología hepatobiliar.
DIAGNÓSTICO DIFERENCIAL Y MANEJO
Ante un paciente con ictericia e hiperbilirrubinemia el objetivo es orientar el proceso diagnóstico en función del tipo de bilirrubina predominante y la presencia o no de datos de gravedad asociados. Es importante detectar las situaciones de insuficiencia hepática (actividad de protrombina <50% que no corrige tras administración de vitamina K) (Figura 1)6.
Figura 1. Algoritmo ictericia. Mostrar/ocultar
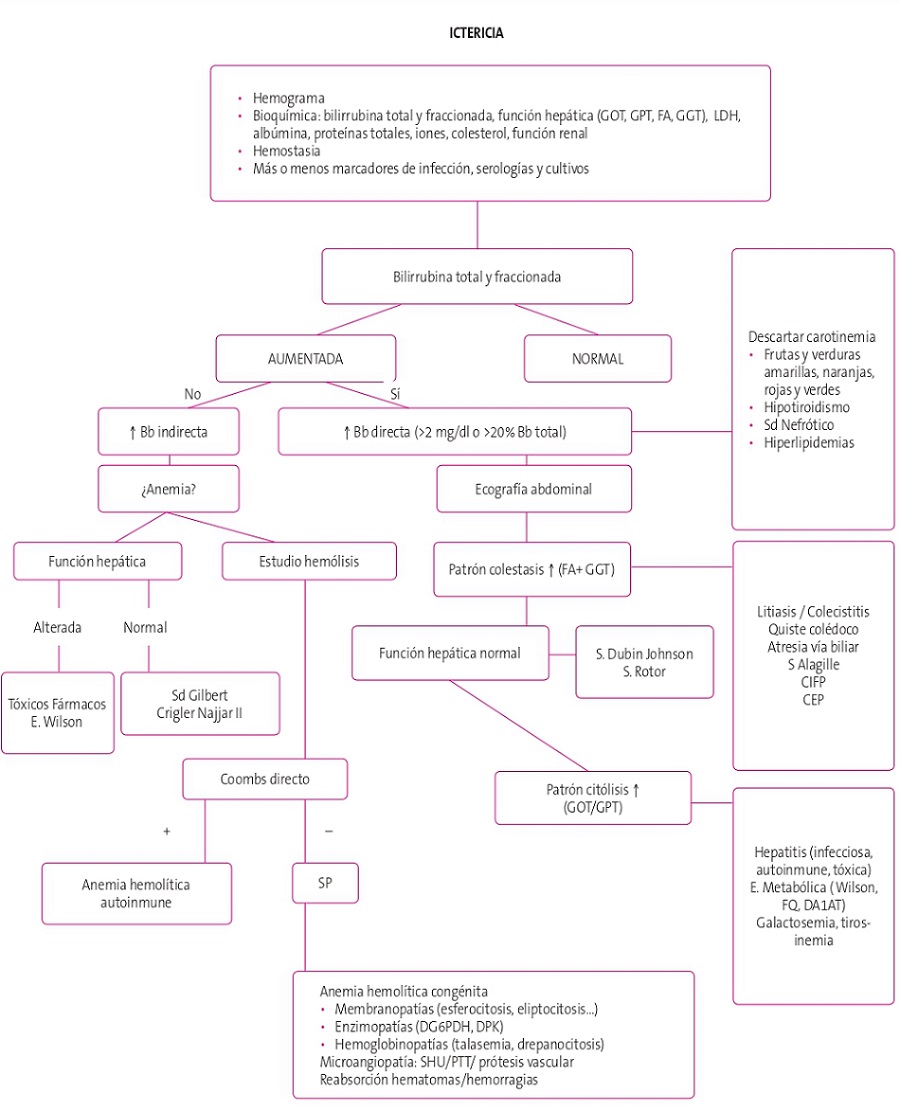
Hiperbilirrubinemia indirecta
La hiperbilirrubinemia indirecta implica el diagnóstico diferencial entre anemia de origen hemolítico, ingesta de medicamentos que alteren la absorción hepática y el síndrome de Gilbert.
En primer lugar, se debe distinguir si existe anemia asociada.
-
Ante valores de hemoglobina normales, se debe evaluar las enzimas hepáticas (ALT/AST).
- Si estas permanecen en rangos de normalidad, el origen de la hiperbilirrubinemia se debe a una alteración de la glucuronidación, siendo la causa más frecuente el síndrome de Gilbert, entidad benigna y más frecuente de hiperbilirrubinemia en adolescentes. Se presenta como episodios de ictericia leve intermitente, en ocasiones desencadenada por ayuno, estrés o enfermedades intercurrentes que no precisa tratamiento. Otra causa infrecuente es la enfermedad de Crigler-Najjar tipo I-II, caracterizada por la ausencia o deficiencia de la enzima UGT. Cursa con hiperbilirrubinemia no conjugada persistente grave en ausencia de hemólisis o enfermedad hepática subyacente que en ocasiones puede precisar tratamiento con fenobarbital.
- El daño hepatocelular manifestado por aumento de las enzimas hepáticas puede deberse a la ingesta de tóxicos o fármacos como la rifampicina, probenecid, etinil-estradiol y gentamicina entre otros, con remisión clínica a las 48 horas de retirada del fármaco.
-
Si existe anemia, se debe completar el estudio analítico con parámetros hemolíticos (descenso de haptoglobina, aumento de LDH, reticulocitosis) y el test de Coombs.
- Un test de Coombs directo positivo es indicativo de anemia hemolítica de origen autoinmune (autoanticuerpos). Los anticuerpos calientes cursan con hemólisis extravascular, siendo la causa idiopática, infecciones o síndromes proliferativos. Por otro lado, los anticuerpos fríos cursan con hemólisis intravascular, siendo la hemoglobinuria paroxística a frigore y las infecciones por Mycoplasma y virus herpes simple las causas más frecuentes.
-
Un resultado negativo en el test de Coombs orienta a anemia hemolítica congénita (membranopatías; esferocitosis, eliptocitosis, enzimopatías; déficit glucosa 6 fosfato deshidrogenasa, hemoglobinopatías; talasemia, drepanocitosis), microangiopatía, hemorragias o reabsorción de hematomas7.
- En las anemias hemolíticas congénitas, la morfología de sangre periférica puede orientar la etiología. La esferocitosis hereditaria es la anemia hemolítica más frecuente en la raza blanca, con herencia autosómica dominante, produciendo un defecto en la membrana del hematíe que ocasiona crisis de hemólisis extravascular. En sangre periférica son característicos los esferocitos, así como un test de fragilidad osmótica alterado. El déficit de glucosa 6 fosfato deshidrogenasa (DG6PD) tiene una herencia ligada al cromosoma X y cursa con crisis hemolíticas ante la exposición a sustancia oxidantes (habas, fármacos o infecciones). Una disminución de la actividad enzimática es diagnóstica de la entidad. La drepanocitosis cursa con crisis vasooclusivas y anemia hemolítica. Es característico la presencia de drepanocitos en frotis de sangre. La talasemia se caracteriza por presentar anemia microcítica y síndrome hemolítico en su forma grave. Ambas hemoglobinopatías se diagnostican mediante electroforesis.
- La presencia de esquistocitos en sangre periférica es característica de anemias microangiopáticas, como el síndrome hemolítico urémico o la púrpura trombocitopénica trombótica.
Hiperbilirrubinemia directa
La hiperbilirrubinemia directa (>2 mg/dl y/o >20% de la Bb total) indica lesión hepatocelular, afectación biliar o patología hereditaria, por lo que se deberá completar el estudio con una prueba de imagen, generalmente ecografía abdominal8,9.
Infección- inflamación hepática
La etiología vírica es la más frecuente. Cuadro clínico de fiebre, ictericia, dolor abdominal, nauseas/vómitos, malestar general, hepatomegalia, aumento de reactantes de infección, enzimas hepáticas (GOT/GPT), y leucocitosis son sugestivos de hepatitis de origen infeccioso. La hepatitis autoinmune cursa en la mitad de los casos con ictericia, dolor abdominal, malestar y vómitos, pudiendo aparecer signos de hepatopatía crónica como hepatoesplenomegalia, arañas vasculares, eritema palmar, circulación colateral si existe retraso diagnóstico. Analíticamente destaca la hipertransaminasemia, pudiendo asociar aumento de GGT, Bb y alteraciones en la coagulación. Los autoanticuerpos ANA/AML, LKM1 son positivos.
Enfermedad biliar extrahepática
Se debe sospechar atresia biliar ante la presencia de ictericia progresiva en las primeras 8 semanas de vida y heces acólicas junto con coluria y hepatomegalia en lactante sano con buen estado general. Es la causa más común de hiperbilirrubinemia conjugada en niños. En la analítica se observa hiperbilirrubinemia directa, aumento de GGT, FA, pudiendo elevarse discretamente las enzimas hepáticas. En la atresia biliar, la ecografía puede mostrar ausencia o hipoplasia de vía biliar extrahepática. Los quistes del colédoco son causa rara de ictericia en la infancia. Se deben descartar ante cuadro clínico de ictericia acompañado de dolor abdominal, nauseas/vómitos, coluria, acolia y patrón analítico de colestasis (aumento GGT, FA, Bb directa) con ligero aumento de transaminasas. En niños mayores la ictericia no siempre está presente.
Enfermedad biliar intrahepática
El síndrome de Alagille se caracteriza por una disminución de conductos biliares intrahepáticos. Destaca la clínica de ictericia, prurito y xantomas, asociando hipocolia, coluria, hepatomegalia y en ocasiones retraso en el desarrollo. En la analítica se objetiva hiperbilirrubinemia directa, aumento GGT, transamisasas e hipercolesterolemia. Asocia facies peculiar con abombamiento frontal, hipertelonerismo y mentón prominente, así como cardiopatía y vértebra en mariposa.
La colestasis intrahepática familiar progresiva (CIFP) debuta a edades tempranas con prurito intenso como síntoma predominante, acompañado de ictericia y hepatomegalia, con hiperbilirrubinemia directa y aumento GGT.
Enfermedad hepatocelular
Las enfermedades metabólicas y genéticas son causa poco frecuente de ictericia en lactantes y niños mayores.
El déficit de alfa-1-antitripsina (menor de 100 mg/dl en sangre) puede incluir en un 10-20% de los casos colestasis debido a la lesión hepatocelular producida por la acumulación de proteína anómala. Se presenta con clínica de ictericia, coluria, acolia, patrón analítico de colestasis sin coagulopatía y suele existir antecedente de bajo peso de recién nacido.
La colestasis infantil es una presentación poco frecuente en pacientes afectos de fibrosis quística.
La tirosinemia se presenta como hepatopatía grave con insuficiencia hepática AP <50% que no es debida a déficit de vitamina K y por tanto no mejora tras administración iv. en los primeros 6 meses de vida. El hallazgo de succinilacetona en orina es diagnóstico.
La galactosemia cursa con aparición de fallo hepático y colestasis de inicio los primeros días de vida tras la ingesta de lactosa.
La hiperbilirrubinemia conjugada aislada sin otras alteraciones en las pruebas hepáticas es característica de dos afecciones hereditarias raras: el síndrome de Dubin-Johnson y el síndrome de Rotor.
CRITERIOS DE INGRESO
Los criterios de ingreso se basan en el estado general y la patología base del paciente. Ingresarán de forma general aquellos pacientes con1:
- Compromiso vital (inestabilidad hemodinámica por anemia, insuficiencia hepática, sepsis, síndrome hemolítico urémico, entre otras causas).
- Mal estado general o necesidad de fluidoterapia intravenosa.
- Crisis hemolíticas.
- Necesidad de tratamiento antibiótico intravenoso (colangitis, colecistitis, sepsis…).
- Insuficiencia hepática aguda (requiere ingreso en unidad de cuidados intensivos pediátricos).
TRATAMIENTO
El manejo terapéutico debe ir orientado en función de la etiología de la hiperbilirrubinemia.
En casos de anemia hemolítica grave y sintomática, puede requerir transfusión de concentrados de hematíes. En la anemia por déficit de glucosa 6 fosfato deshidrogenasa se debe evitar la exposición a sustancias que desencadenen crisis hemolíticas como las habas, fármacos antipalúdicos, ácido acetil salicílico, nitrofurantoína, entre otros. Si el origen hemolítico es autoinmune, no se debe administrar corticoterapia hasta realizar un estudio etiológico, ya que estos son efectivos únicamente ante anticuerpos calientes.
Los quelantes e inductores enzimáticos como ácido ursodesoxicólico, fenobarbital y rifampicina entre otros, son parte del tratamiento del síndrome colestásico.
Se debe valorar tratamiento quirúrgico en caso de quiste de colédoco, atresia de vías biliares, litiasis u otras causas de ictericia obstructiva.
Si existe coagulopatía asociada, se debe administración de vitamina K.
CUADERNO DEL PEDIATRA
- La ictericia fuera del periodo neonatal es poco frecuente.
- Ante todo paciente con ictericia, se debe determinar la bilirrubina total y directa.
- Se añadirán pruebas analíticas y/o de imagen en función de la fracción de bilirrubina elevada predominante.
- La mayoría de los casos requerirán ingreso para completar el estudio etiológico.
- El tratamiento dependerá de la patología subyacente.
BIBLIOGRAFÍA
- Guerrero-Fernández J, Cartón Sánchez A, Barreda Bonis A, Menéndez Suso J, Ruiz Domínguez J. Ictericia no neonatal. En: Manual de diagnóstico y terapéutica en Pediatría. Madrid: Editorial Médica Panamericana; 2018. p. 205–10.
- Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, et al. Guideline for the evaluation of cholestatic jaundice in infants: Joint recommendations of the North American society for pediatric gastroenterology, hepatology, and nutrition and the European society for pediatric gastroenterology, hepatology, and nutrition. J Pediat Gastroent Nutr. 2017; 64:154-68.
- Roy-Chowdhury N, Roy-Chowdhury J. Bilirubin metabolism. En: Arias IM, Alter HJ, Boyer JL, Cohen DE, Shafritz DA, Thorgeirsson SS, Wolkoff AW (ed.). The Liver: Biology and Pathobiology. Wiley; 2022.
- Ronald J, Wong B, Vinod K Bhutani MF. Unconjugated hyperbilirubinemia in the newborn: Pathogenesis and etiology. En: UpToDate [en línea] [consultado el 15/11/2022]. Disponible en https://www.medilib.ir/uptodate/show/5020.
- Martínez Velasco S, de la Calle Navarro E, Tutau Gómez C. Ictericia. En: Díaz González L, Mesa Fumero Y (ed.). Urgencias pediátricas. Guía de actuación. 2.ª edición. Madrid: Ergon; 2020. p. 573–9.
- Vigil S, Lorente J, Míguez C. Protocolo de actuación ante ictericia en el lactante y niño mayor en urgencias. Hospital General Universitario Gregorio Marañón. Revisión junio 2017. En: Comunidad de Madrid [en línea] [consultado el 15/11/2022]. Disponible en https://www.comunidad.madrid/hospital/gregoriomaranon/file/3437/download?token=2hYyqhqy.
- Sáez González I, Daghoum Dorado E. Anemia. En: Díaz L, Mesa Y (ed.). Urgencias pediátricas. Guía de actuación. 2.ª edición. Madrid: Ergon; 2019. p. 783–98.
- Guerrero-Fernández J, Cartón Sánchez A, Barreda Bonis A, Menéndez Suso J, Ruiz Domínguez J. Colestasis. En: Manual de diagnóstico y terapéutica en Pediatría. Madrid: Editorial Médica Panamericana; 2018. p. 1029–42.
- Guerrero-Fernández J, Cartón Sánchez A, Barreda Bonis A, Menéndez Suso J, Ruiz Domínguez J. Disfunción hepática. En: Manual de diagnóstico y terapéutica en Pediatría. Madrid: Editorial Médica Panamericana; 2018. p. 999–1028.
LECTURAS RECOMENDADA
- Guerrero-Fernández J, Cartón Sánchez A, Barreda Bonis A, Menéndez Suso J, Ruiz Domínguez J. Ictericia no neonatal. En: Manual de diagnóstico y terapéutica en Pediatría. Madrid: Editorial Médica Panamericana; 2018. p. 205–10.
- Martínez Velasco S, de la Calle Navarro E, Tutau Gómez C. Ictericia. En: Díaz González L, Mesa Fumero Y (ed.). Urgencias pediátricas. Guía de actuación. 2.ª edición. Madrid: Ergon; 2020. p. 573–9.
- Vigil S, Lorente J, Míguez C. Protocolo de actuación ante ictericia en el lactante y niño mayor en urgencias. Hospital General Universitario Gregorio Marañón. Revisión junio 2017. En: Comunidad de Madrid [en línea] [consultado el 15/11/2022]. Disponible en: https://www.comunidad.madrid/hospital/gregoriomaranon/file/3437/download?token=2hYyqhqy.