


Oncología para el pediatra de Atención Primaria (II): formas de presentación de las diferentes neoplasias infantiles
RESUMEN: PUNTOS CLAVE PARA EL PEDIATRA DE ATENCIÓN PRIMARIA
SUMMARY: KEY POINTS FOR PRIMARY CARE PEDIATRICIANS
PUNTOS CLAVE
- La patología neoplásica infantil ocupa un pequeño espacio dentro de las afecciones pediátricas, pero su identificación precoz tiene importantes implicaciones pronósticas y terapéuticas.
- El diagnóstico precoz se basa en tres pilares: alto índice de sospecha, identificación y seguimiento de pacientes de riesgo y detección precoz de signos/síntomas de alarma.
- Debe darse valor a los cambios bruscos comportamentales referidos por los padres en el contexto de síntomas comunes prolongados en el tiempo, de curso atípico o asociados a síndrome constitucional o alteraciones sugerentes de malignidad en la exploración física.
- En presencia de cefalea de curso menor a seis meses, en niños menores de tres años, de duración prolongada con empeoramiento progresivo, aparición matutina, despertares nocturnos, asociación a vómitos, focalidad neurológica y/o alteración oftalmológica, debe descartarse una lesión ocupante de espacio intracraneal.
- En caso de adenomegalias persistentes (4-6 semanas), rápidamente progresivas, >2,5 cm sin respuesta a antibióticos, de localización supraclavicular, retroauricular o epitroclear con características patológicas, asociadas a alteraciones hematológicas, radiológicas o hepatoesplenomegalia y/o síndrome constitucional, debe descartarse malignidad.
- En dolores osteoarticulares multifocales, progresivos, de aparición nocturna y con mala respuesta analgésica, sin antecedente traumático, con palpación de masa de partes blandas y/o manifestaciones sistémicas, debe descartarse neoplasia sólida o hematológica.
- La fiebre prolongada (>14 días) de origen desconocido junto con síndrome constitucional, palidez, púrpura, adenopatías y/o hepatoesplenomegalia, podría tener etiología neoplásica.
- En caso de sospecha de cáncer infantil, el pediatra de Atención Primaria debe orientar e iniciar el estudio diagnóstico, derivando de forma precoz al niño a un centro con Servicio de Oncología y Hematología Pediátricas. Es preferible derivar a servicios médicos que quirúrgicos, pues estos se encargarán de la coordinación con el resto de especialistas.
- Existen verdaderas urgencias oncológicas que deben ser derivadas sin demora, como el síndrome linfoproliferativo con lisis tumoral, las masas mediastínicas o abdominales, el síndrome de vena cava superior, el síndrome de compresión medular y los tumores cerebrales con signos de hipertensión intracraneal.
FORMAS DE PRESENTACIÓN DE LAS DIFERENTES NEOPLASIAS INFANTILES
Leucemia
Es la neoplasia más frecuente en la infancia, representa un 25-30% de los casos en menores de 14 años1-3. Es más frecuente entre uno y diez años de edad, en varones y en la raza blanca4. Más del 95% de las leucemias en la infancia son agudas, y entre ellas predomina la linfoblástica3. Es habitual que la presentación tenga un curso insidioso, subagudo, de semanas de evolución. La presencia de un cuadro febril prolongado (más de dos semanas) o recurrente, junto con la presencia de linfadenopatías generalizadas, hepatoesplenomegalia, astenia, anorexia, pérdida de peso, irritabilidad, dolores osteoarticulares, impotencia funcional y/o alteraciones de la marcha, debe hacer pensar al pediatra en una leucemia, si bien estos hallazgos se observan también en procesos benignos (por ejemplo, infecciones virales)3,5-7. Debe prestarse especial atención a los cambios de carácter o de comportamiento referidos por los padres8.
La anemia secundaria a la infiltración medular produce palidez, astenia, soplo cardiaco y/o taquicardia, así como acúfenos, cefalea, vértigo, disnea e insuficiencia cardiaca en casos más graves. La trombocitopenia se manifiesta con epistaxis, gingivorragia, petequias, equimosis, hematomas (generalizados o en localizaciones atípicas, a veces confundidos con un maltrato infantil), hematuria y/o melenas2,3,9. Puede existir alteración de la coagulación, con diátesis hemorrágica, especialmente en la leucemia promielocítica (M3). La neutropenia puede conducir a infecciones graves (gérmenes atípicos, evolución tórpida), mucositis y aftas orales. En caso de afectación del sistema nervioso aparecería cefalea, afectación de pares craneales (especialmente III, IV, VI y VII), hemorragia intracraneal, síndrome meníngeo, afectación de médula espinal y/o convulsiones. En varones, puede observase infiltración testicular (aumento de volumen). En leucemias mieloides son características las lesiones orales con hiperplasia gingival, los cloromas (sarcoma granulocítico) y los nódulos subcutáneos azulados-verdosos(blueberry muffin), en especial en las monocíticas (M4, M5). En la práctica, cualquier órgano puede ser infiltrado por blastos leucémicos, produciendo nefromegalia, masa mediastínica (leucemias T), infiltrados pulmonares, derrame pericárdico, afectación de glándulas3...
Los hallazgos anteriores deben considerarse especialmente en niños con predisposición genética a desarrollar leucemia, esto es, los que presentan alguna alteración cromosómica o síndrome genético predisponente (síndrome de Down, anemia de Fanconi, neurofibromatosis, ataxia-telangiectasia, Bloom, Wiskott-Aldrich, Turner o Poland, inmunodeficiencias, etc.). Además, en hermanos de sujetos afectos de leucemia se ha descrito un riesgo entre dos y cuatro veces mayor que en la población general2. En gemelos homocigotos, el riesgo en el hermano sano se eleva hasta un 20-25%, y es aún mayor si la leucemia aconteció el primer año de vida2.
Si sospechamos una leucemia debemos realizar un hemograma urgente con reticulocitos y con frotis de sangre periférica, una bioquímica con parámetros de lisis tumoral, coagulación y reactantes de fase aguda. Es típica una anemia normocítica normocrómica, hiporregenerativa (reticulocitopenia), trombopenia, linfocitosis o leucocitosis, neutropenia, hipereosinofilia y blastos o células atípicas en el frotis. Si existe lisis tumoral, observaremos un aumento de los valores séricos de ácido úrico, potasio, fósforo y lactatodeshidrogenasa. En leucemias promielocíticas, puede observarse coagulopatía intravascular diseminada. En caso de que se realice una radiografía simple por los dolores óseos referidos, es habitual observar una osteopenia difusa, lesiones osteolíticas, osteosclerosis y bandas transversas radiotransparentes. En la radiografía de tórax puede observarse un ensanchamiento mediastínico, especialmente en leucemias/linfomas T3.
Linfomas no Hodgkin
Suponen aproximadamente un 7-8% de los tumores malignos infantiles. Son tumores de rápido crecimiento, generalmente agresivos en su presentación, localizados en cualquier ubicación donde haya tejido linfoide. Pueden asociarse a antecedentes infecciosos (virus de la inmunodeficiencia humana, Epstein-Barr)10. La sintomatología inicial puede ser inespecífica (tos, odinofagia, dolor abdominal, vómitos...), pero la rápida progresión suele orientar hacia un proceso linfoproliferativo maligno: síndrome constitucional, aumento de volumen de ganglios linfáticos, masa cervical (a veces con parálisis de pares craneales u obstrucción nasal), masa mediastínica (insuficiencia respiratoria, síndrome de vena cava superior, derrame pleural), masa abdominal (distensión, dolor abdominal, abdomen agudo, estreñimiento, invaginación intestinal, obstrucción intestinal), asimetría amigdalar o dolor mandibular (por infiltración)9,10. En cualquier niño mayor de seis años con una invaginación intestinal debe considerarse la posibilidad de linfoma en el diagnóstico diferencial5. Pueden afectar al sistema nervioso central o a la médula ósea por infiltración. En ellos, especialmente en los de tipo Burkitt, existe un elevado riesgo de producir un síndrome de lisis tumoral. Un estudio analítico inicial puede ayudar al diagnóstico (hemograma, velocidad de sedimentación globular, marcadores de lisis tumoral), así como estudios básicos de imagen (radiografía de tórax, ecografía de partes blandas).
Linfomas de Hodgkin
Aparecen de forma predominante en varones adolescentes, siendo raros antes de los diez años de vida. Suponen un 6% de los tumores malignos infantiles. Se caracterizan por un aumento significativo del volumen de los ganglios linfáticos, generalmente supraclaviculares y/o cervicales (también mediastínicos, axilares, paraaórticos e iliacos) y puede existir afectación esplénica, del hueso, pulmonar o de la médula ósea (raramente). En algunos pacientes se manifiesta como tos persistente por la masa mediastínica5,10. Las adenopatías se caracterizan por ser indoloras, experimentando un crecimiento lento pero progresivo con características patológicas (duras, fijas, adheridas a planos profundos). En aproximadamente un tercio de los pacientes se asocia sintomatología inespecífica: síntomas B (fiebre de origen inexplicable, de aparición intermitente y recurrente, sudoración nocturna y pérdida de peso involuntaria, más de un 10% en menos de seis meses), prurito, cansancio, somnolencia o anorexia9,10.
Toda masa no dolorosa, especialmente cervical, que no responde a antibióticos debe ser investigada mediante ecografía y radiografía de tórax5. Ante la sospecha de un linfoma, el paciente debe ser derivado de forma urgente a un centro hospitalario para completar el estudio diagnóstico (resonancia magnética, tomografía por emisión de positrones-tomografía computarizada (PET-TAC), biopsia ganglionar...).
Tumores del sistema nervioso central
Representan el segundo tumor en frecuencia en la infancia, en torno al 20% de los tumores de la edad pediátrica1. Los síntomas suelen ser vagos y poco específicos inicialmente, y en muchas ocasiones no contamos con la colaboración del niño para describir su sintomatología. La clínica aparece relacionada con la hipertensión intracraneal que el tumor produce en su crecimiento o bien con la compresión-infiltración de las estructuras adyacentes al mismo, según su localización (Tabla 1)9,10. En caso de sospecha de hipertensión intracraneal, acompañada o no de déficit focal, es mandatorio realizar una exploración del fondo de ojo.
Tabla 1. Signos y síntomas sugerentes de un tumor del sistema nervioso central Mostrar/ocultar

Wilne S.et al. realizaron un metaanálisis11 en el que se recogía la semiología descrita al diagnóstico en un total de 4171 pacientes pediátricos diagnosticados de un tumor del sistema nervioso central. Los síntomas y signos más frecuentes en el diagnóstico fueron2,11: cefalea (33%), náuseas y vómitos (32%), alteraciones de la marcha/coordinación (27%) y edema de papila (13%), en tumores intracraneales; macrocefalia (41%), náuseas y vómitos (30%), irritabilidad (24%) y letargo (21%), en niños menores de cuatro años con tumores intracraneales; disminución de la agudeza visual (41%), exoftalmia (16%) y atrofia óptica (15%), en pacientes con antecedente de neurofibromatosis; náuseas y vómitos (75%), cefalea (67%), trastornos de la marcha/coordinación (60%) y edema de papila (34%), en tumores de fosa posterior; signos de hipertensión intracraneal (47%), convulsiones (38%) y edema de papila (21%), en tumores supratentoriales; trastornos de la marcha y la coordinación (78%), parálisis de los nervios craneales (52%), signos piramidales (33%), cefalea (23%) y estrabismo (19%), en tumores del tronco cerebral; dolor de espalda (67%), alteraciones de la marcha/coordinación (42%), deformidad de la columna (39%), debilidad focal (21%) y pérdida de control de esfínteres (20%), en tumores medulares. Si su origen es hipotálamo-hipofisario aparecen deficiencias visuales, panhipopituitarismo, diabetes insípida y/o trastornos de la pubertad. Los signos de hipertensión intracraneal se ausentan en más de la mitad de los niños con un tumor cerebral.
La sospecha debe ser elevada si se asocia más de un signo o síntoma sugerente de lesión intracraneal ocupante de espacio (Tabla 1), en particular si son progresivos. En pacientes afectos de neurofibromatosis, esclerosis tuberosa, von Hippel-Lindau, ataxia-telangiectasia o síndrome de Gorlin debemos tener un alto índice de sospecha ante sintomatología sugerente2.
Neuroblastoma y otros tumores del sistema nervioso simpático
Es la neoplasia sólida extracraneal más frecuente de la infancia, en torno a un 8,8% de los casos de cáncer1 y un 15% de las neoplasias infantiles diagnosticadas por debajo de los cinco años. Es característico de la infancia temprana, un 80% de los casos se diagnostican antes de los cuatro años de vida (un tercio de ellos antes de los dos años)10. Se origina a partir de la glándula suprarrenal o de los ganglios simpáticos paravertebrales, por lo puede aparecer a lo largo de todo el eje espinal. Su localización más frecuente es abdominal (área retroperitoneal y glándula suprarrenal, 65-69%), seguida del mediastino posterior (20%), la región cervical (5%) y la pelvis (2-3%)9.
Las manifestaciones clínicas son frecuentemente inespecíficas (fiebre, pérdida de peso, astenia, hiporexia, estancamiento ponderal, adenopatías, dolores osteoarticulares, claudicación de la marcha...). Aparecen síntomas en relación con el efecto masa (tos persistente, disnea, distensión abdominal, estreñimiento, obstrucción intestinal) o por la compresión medular o de raíces nerviosas (dolor de espalada, de características radiculares con paraplejia, incontinencia urinaria y/o fecal)9,12. De forma poco frecuente, presenta hipertensión arterial, habitualmente por compresión de la arteria renal (no por la secreción de catecolaminas, más típica del feocromocitoma). Se diferencian distintos síndromes que, solos o en combinación, se asocian al debut del neuroblastoma: 1)síndrome de Pepper (hepatomegalia por infiltración masiva del hígado), en formas metastásicas en recién nacidos y lactantes; 2) síndrome de Horner (ptosis, miosis y anhidrosis), en tumores cervicales o torácicos; 3) síndrome de vena cava superior en tumores cervicales e intratorácicos; 4) síndrome de Hutchinson (hematoma en “anteojos” o en “ojos de mapache”, con proptosis), en caso de infiltración retrobulbar y orbitaria; a veces se manifiesta como dolor óseo, anemia, hemorragia y/o infecciones, si hay infiltración de la médula ósea; y 5) síndrome deblueberry muffin (nódulos subcutáneos azulados por invasión cutánea), característico de neonatos y lactantes.
El neuroblastoma es un tumor característicamente asociado a síndromes paraneoplásicos12: 1) síndrome opsoclonus-mioclonus: caracterizado por la aparición de movimientos oculares involuntarios, caóticos, conjugados, de gran amplitud, asociados a mioclonías focales y ataxia de tronco; 2) síndrome de Kerner-Morrison (7-9%): diarrea secretora, hipocaliemia y deshidratación, por secreción de péptido vasoactivo intestinal (VIP) y somatostatina, en formas más benignas (ganglioneuromas, ganglioneuroblastomas), de mejor pronóstico; 3) hipercalcemia de origen tumoralpor secreción de moléculas similares a la paratohormona.
Tumor de Wilms (nefroblastoma)
Es el tumor renal más frecuente en niños, en torno a un 5%. Debuta habitualmente entre uno y cinco años de edad, con un pico de incidencia a los 2-3 años2. En un 75% de los casos se descubre como un hallazgo casual de una masa abdominal. Su localización es anterior y suele circunscribirse a un hemiabdomen. Un 25% asocia hipertensión arterial y/o hematuria. Es habitual el antecedente de dolor abdominal, vómitos, pérdida del apetito, estreñimiento, fiebre o infección del tracto urinario9,10. Se presenta de forma bilateral en un 3-13% de los casos.
El tumor de Wilms es el ejemplo clásico de neoplasia asociada a trastornos genéticos (solo un 1-4%)2,12, pero debe tenerse en consideración para realizar un seguimiento estrecho. En sujetos afectos de síndrome WARG o síndrome de Denys-Drash existe un 33-50% de riesgo de desarrollar un nefroblastoma. Su incidencia en niños con aniridia es del 4,2-5,5%; con hemihipertrofia, de un 5,8-15,9%, y con síndrome de Beckwith-Wiedemann, de un 0,8-1,9%13.
Sarcoma de partes blandas
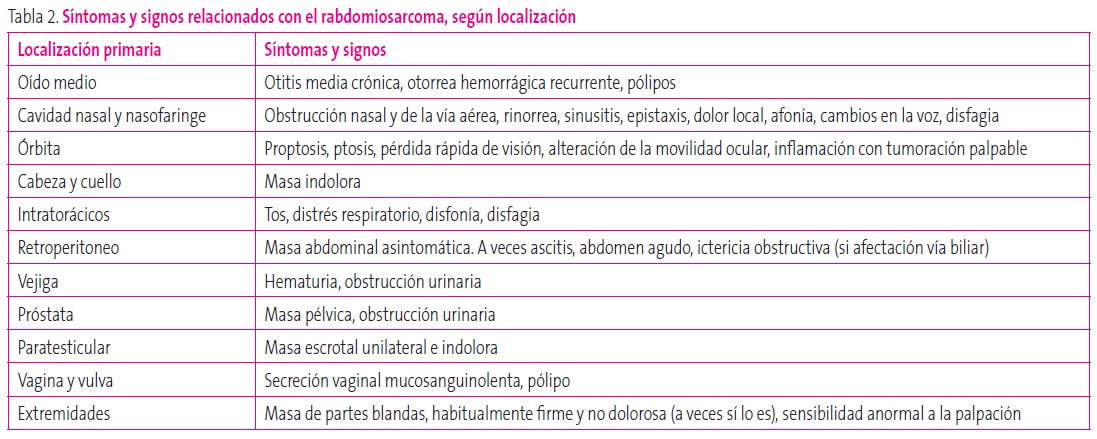
Los sarcomas de partes blandas suponen un 6,3% de los tumores infantiles, siendo el más frecuente el rabdomiosarcoma. Existen dos picos de incidencia (2-6 años y adolescencia), con un ligero predominio en varones. Su variante histológica embrionaria es más común en niños pequeños y lactantes. El tipo alveolar, más agresivo, de localización preferente en tronco y extremidades, aparece sobre todo en niños mayores de seis años. Se asocia con alteraciones citogenéticas y con formas hereditarias (síndrome de Li-Fraumeni o neurofibromatosis)10. Su localización es muy diversa: el tipo embrionario aparece preponderantemente en la cara –nasofaringe, cavidad nasal, conducto auditivo, región mastoidea, parótida–, la pelvis, las vías biliares y el hígado, y la región paratesticular9; el tipo alveolar se localiza habitualmente en las extremidades, la pared del tronco, en localización intratorácica o intraabdominal9. Según la localización, podemos encontrar diversa sintomatología, que se desarrolla en la Tabla 2.
Tabla 2. Síntomas y signos relacionados con el rabdomiosarcoma, según localización Mostrar/ocultar
Tumores de estirpe ósea
Los tumores óseos malignos suponen un 5,5% de las neoplasias infantiles. Se observan con mayor frecuencia en la segunda década de la vida, coincidiendo con el estirón puberal2, aunque los tumores de la familia Ewing pueden verse en niños más pequeños. El dolor localizado en los huesos (especialmente en el fémur) o en las articulaciones (sobre todo en la rodilla) es un motivo de consulta habitual en Pediatría.
Los tumores óseos son raros, pero debemos pensar en ellos especialmente en la segunda década de la vida y si no existe antecedente traumático previo5. Suelen presentarse con dolor óseo localizado (a veces multifocal), inicialmente intermitente (que se hace persistente), que no calma con analgésicos y desvela por la noche, acompañado posteriormente de una masa palpable (tumoración de consistencia dura) y de una alteración de la funcionalidad9. En el caso del sarcoma de Ewing, aparece fiebre en un cuarto de los pacientes. A veces se observa pérdida de peso. En el estudio radiológico puede observarse una fractura patológica con un aumento de partes blandas que no es proporcional a la lesión9. El sarcoma de Ewing muestra clásicamente un patrón de múltiples láminas (“capas de cebolla”). El osteosarcoma presenta áreas osteoclásticas y osteoblásticas dentro del tumor, siendo típico el triángulo de Codman (formación de hueso reactivo entre el periostio intacto elevado y la cortical subyacente en la zona de transición con la zona extraósea del tumor)10. Es frecuente que exista un retraso entre el inicio de los síntomas y el diagnóstico de estos tumores, especialmente en localizaciones axiales o pélvicas, debido a lo insidioso del curso de la enfermedad.
Retinoblastoma
Es el tumor ocular más frecuente en niños, en torno a un 3% del global. Un 90% aparece en menores de cuatro años (en especial durante el primer año de vida). Existe una forma hereditaria, a menudo de presentación bilateral y multifocal, con una herencia autosómica dominante y elevada penetrancia. Los hallazgos físicos que nos deben alarmar son leucocoria (reflejo pupilar blanco), estrabismo de reciente aparición (sobre todo en mayores de tres meses), inflamación ocular persistente, proptosis, disminución de la agudeza visual y glaucoma. Es importante conocer los antecedentes familiares con objeto de discriminar las formas hereditarias de las esporádicas. Parece existir una asociación entre el retinoblastoma con el labio leporino, la dentinogénesis imperfecta, la incontinencia pigmenti y la catarata familiar congénita2.
Hepatoblastoma
Supone un 0,6-1% de los tumores malignos infantiles. Se presenta en general con hepatomegalia acompañada o no de otros síntomas: distensión abdominal, ictericia, pérdida de peso, anorexia, vómitos, fiebre o dolor abdominal. Sus metástasis suelen localizarse en el pulmón y, menos frecuentemente, en el hueso, el sistema nervioso central y la médula ósea10.
Tumores germinales
Suponen un 3,5% de los casos de neoplasias infantiles1. Aparecen en múltiples localizaciones, de modo que la clínica de presentación dependerá de su ubicación10,14:
- Región sacrococcígea (42%): se asocia con frecuencia a anomalías congénitas (especialmente musculoesqueléticas o del sistema nervioso), y son más frecuentes en niñas (75%). A menudo se diagnostican como hallazgo casual en una exploración de imagen realizada por otro motivo o en relación con la palpación de una masa abdominal o una masa exofítica en la región sacrococcígea.
- Ovario (24%): se presentan generalmente a partir de los ocho años de vida, especialmente entre los 10 y los 14 años, con un pico de aparición a los 18-19 años de edad. Se manifiestan con dolor abdominal (generalmente crónico, aunque en un tercio simulan un abdomen agudo en relación con torsión ovárica), masa abdominal, fiebre recurrente, estreñimiento, amenorrea, sangrado vaginal, pubertad precoz y, raramente, disuria.
- Testículo (9%): existe una importante asociación con antecedentes de criptorquidia. A menudo su presentación es paucisintomática, en forma de masa escrotal indolente. Raramente se presenta como pubertad precoz.
- Mediastino (8%): se presentan casi siempre como una masa mediastínica anterior, más frecuentes en varones, y producen disnea, insuficiencia respiratoria, hemoptisis…
- Intracraneales:están localizados generalmente en la región pineal o supraselar. La presentación incluye alteraciones visuales, diabetes insípida, hipopituitarismo, anorexia y pubertad precoz.
- Otras localizaciones (retroperitoneo, cuello, vagina…).
El diagnóstico se basa en la realización de pruebas de imagen pertinentes según la localización, así como de la realización de marcadores tumorales (AFP, β-HCG) y lactato deshidrogenasa.
Histiocitosis
Se trata de un grupo heterogéneo de enfermedades de causa desconocida, muy infrecuentes, a menudo infradiagnosticadas, caracterizadas por una proliferación de células del sistema mononuclear-fagocítico, con gravedad variable. Puede presentarse en cualquier rango etario, pero son más frecuentes entre uno y tres años de edad, con una incidencia estimada de 3-5 casos/106 niños/año16. El subtipo más relevante en Pediatría es la histiocitosis de células de Langerhans. Puede aparecer de forma localizada o multisistémica, con afectación de dos o más órganos: hueso, mucosas, piel, médula ósea, ganglios linfáticos, sistema nervioso (eje hipotálamo-hipofisario) o vísceras (hígado, pulmón, bazo…)10. En raras ocasiones (<2%) afecta al aparato digestivo, en forma de diarrea persistente o síndrome malabsortivo15.
En lactantes y niños pequeños, se presenta de forma típica en forma de lesiones cutáneas, eritematocostrosas, con coloración roja-violácea, aisladas o confluentes, a menudo con descamación superficial y un curso tórpido, pudiendo presentar periodos de regresión parcial. Pueden simular otros procesos como dermatitis seborreicas. Suelen localizarse en el cuero cabelludo, la región retroauricular, el tronco, los pliegues y, raramente, en las palmas o las plantas de los pies. Las formas multisistémicas son más frecuentes en niños menores de tres años, lo que implica un peor pronóstico15.
En niños mayores de 2-3 años es frecuente encontrar sintomatología relacionada con afectación ósea, por orden de frecuencia, en cráneo, huesos largos, huesos planos y vértebras. Se consideran de riesgo las afectaciones hematopoyética, hepática, esplénica y pulmonar, así como determinadas localizaciones óseas, a saber, en vértebras, órbita, temporal, mastoides, esfenoides, arco zigomático, etmoides, maxilar y/o senos paranasales y fosa craneal media o anterior, con extensión a tejidos blandos. Puede presentarse con afectación ocular (proptosis, exoftalmos, afectación orbitaria), auricular (otitis externa/media recurrente, otorrea persistente) o de cavidad oral (movilidad o pérdida de piezas dentarias, masa intraoral, úlcera mucosa, gingivitis)15,16. En caso de afectación del sistema nervioso central, en especial del hipotálamo, se manifiesta con diabetes insípida con polidipsia y poliuria, y panhipopituitarismo.
El diagnóstico se basa, como siempre, en una adecuada anamnesis y exploración física, siendo en este caso fundamental la histopatología característica de estas lesiones (CD1a, CD207), en caso de poder realizar una biopsia. El estudio de extensión consiste en un hemograma con extensión de sangre periférica (y estudio de médula ósea si existen citopenias), bioquímica de sangre y orina con osmolalidad e iones (para descartar una diabetes insípida), función hepática, ecografía abdominal, radiografía de tórax y serie ósea. En casos en que se sospecha afectación multifocal, se indicarán otras pruebas complementarias (resonancia magnética selectiva o total body, gammagrafía ósea, PET…).
EXÁMENES COMPLEMENTARIOS DE PRIMERA LÍNEA
Existe una gran variedad de estudios analíticos, anatomopatológicos, moleculares y de imagen que nos pueden ayudar a diagnosticar una neoplasia infantil. De forma breve, las herramientas diagnósticas más habituales en la práctica de Atención Primaria son10:
- Hemograma, velocidad de sedimentación globular, bioquímica con perfil hepático y renal así como marcadores de lisis tumoral (ácido úrico, lactatodeshidrogenasa, iones),fosfatasa alcalina, proteinograma y serologías virales o parasitológicas (por ejemplo, Leishmania).
-
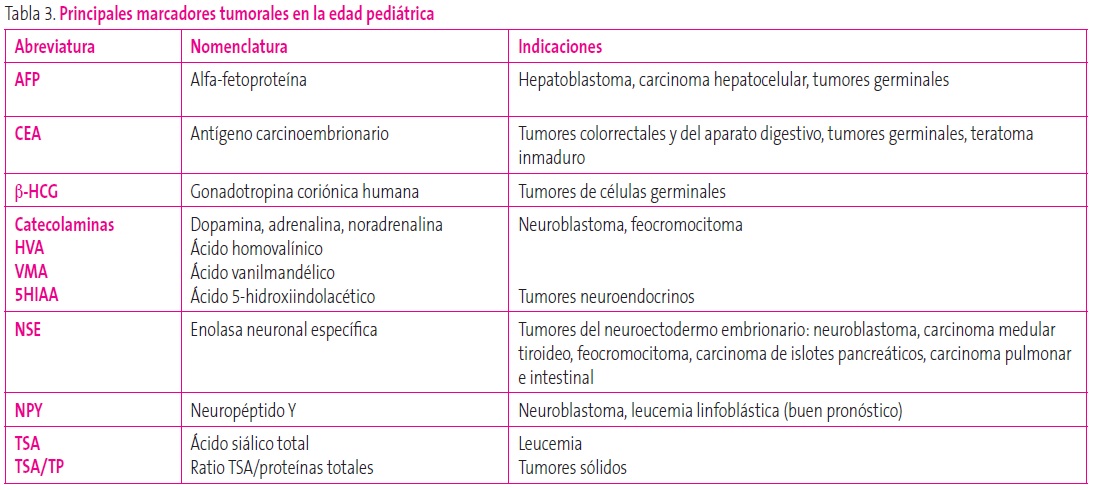
Marcadores tumorales: en la práctica clínica, los más relevantes son los metabolitos de catecolaminas en orina y la alfafetoproteína, pero hay muchos otros (Tabla 3)17.
Tabla 3. Principales marcadores tumorales en la edad pediátrica Mostrar/ocultar
-
Estudios de imagen:
- Radiografía de tórax: debe hacerse anteroposterior y lateral cuando sospechemos tumoraciones intratorácicas primarias o metastásicas, mediastínicas, derrame pleural maligno o tumores cardiacos.
- Radiografía de abdomen: su indicación principal es la sospecha de una obstrucción intestinal derivada de una masa abdominal o pélvica. No obstante, hay que sopesar su uso, pues una radiografía normal no descarta una enfermedad neoplásica abdominal, pero supone una dosis de radiación para el niño.
- Ecografía abdominal: es el estudio de elección ante cualquier sospecha de masa abdominal o pélvica, y resulta especialmente útil para conocer las dimensiones, características y relaciones de vecindad con tejidos adyacentes. El estudio Doppler nos daría información sobre vascularización de la masa.
- Otros estudios de imagen más avanzados se reservarán para el medio hospitalario: resonancia magnética nuclear, tomografía computarizada, PET-TAC, tomografía computarizada por emisión de fotón simple ósea, gammagrafía (en sus diferentes modalidades)...
El resto de estudios hematológicos (por ejemplo, aspirado de médula ósea), anatomopatológicos, citogenéticos, de biología molecular, etc. se harán en medio hospitalario en función de la sospecha y de los datos arrojados previamente por los estudios de su pediatra de referencia.
BIBLIOGRAFÍA
- Registro Nacional de Tumores Infantiles de la Sociedad Española de Hematología y Oncología Pediátricas (RNTI-SEHOP) [base de datos en Internet]. Valencia: Universitat de València,2014 [en línea]. Disponible en: http://www.uv.es/rnti/pdfs/B1.05-Texto.pdf
- García Hernández B. Signos y síntomas sugerentes de cáncer en la infancia en Atención Primaria. Pediatr Integral. 2004.VIII:524-32.
- García Bernal M, Badell Serra I. Leucemia en la infancia: signos de alerta. An Pediatr Contin. 2012;10:1-7.
- Fernández-Teijeiro Álvarez A. Signos de sospecha de neoplasias. En: XXII Congreso Nacional de la Sociedad Española de Pediatría Extrahospitalaria y de Atención Primaria. Tenerife, octubre de 2008.
- Young G, Toretsky JA, Campbell AB, Eskenazi AE. Recognition of common childhood malignancies. Am Fam Physician. 2000;61:2144-54
- Fragkandrea I, Nixon JA, Panagopoulou P. Signs and symptoms of childhood cancer: a guide for early recognition. Am Fam Physician. 2013;88:185-92
- Pession A. Segni di allarme per la diagnosiprecoce di leukemiaacuta. Minerva Pediatr. 2009;61:845-7.
- Clarke RT, Jones CH, Mitchell CD, Thompson MJ. 'Shouting from the roof tops': a qualitative study of how children with leukaemia are diagnosed in primary care. BMJ Open. 2014;4:e004640
- García Hernández B. Sospecha de cáncer en pediatría. Pediatr Integral 2008;XII:537-44.
- Álvarez Silván AM, Santana V. Signos y síntomas de alarma en el cáncer infantil. Oncopedia – Cure4kidsTM; 2010.
- Wilne S, Collier J, Kennedy C, Koller K, Grundy R, Walker D. Presentation of childhood CNS tumours: a systematic review and meta-analysis. Lancet Oncol. 2007;8:685-95
- Pachecho Cumani M. Tumores de cresta neural. Pediatr Integral 2004;VIII:489-98.
- Rodrigues KE, De Camargo B. Diagnóstico precoce do câncer infantil: responsabilidade de todos. Rev Assoc Med Bras. 2003;49:29-34
- Cushing B, Perlman EJ, Marina NM, Castleberry RP. Chapter 36. Germ cell tumors. En: Pizzo PA, David G. Poplack. Principles and Practice of Pediatric Oncology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2011:1118-36.
- McClain KL. Clinical manifestations, pathologic features, and diagnosis of Langerhans cell histiocytosis. 2014. En UpToDate [en línea] [consultado el 10/06/2014]. Disponible en: http://goo.gl/sGLiqM
- Minkow M, Grois N, McClain K, Nanduri V, Rodriguez-Galindo C, Simonitsch-Klupp I. Langerhans cell histiocytosis. Histiocyte Society Evaluation and Treatment Guidelines; April 2009.
- Lahdenne P, Heikinheimo M. Clinical use of tumor markers in childhood malignancies. Ann Med. 2002;34:316-23
Cómo citar este artículo
Artículos relacionados
 Oncología para el pediatra de Atención Primaria (I): signos y síntomas sugerentes de patología neoplásica
Oncología para el pediatra de Atención Primaria (I): signos y síntomas sugerentes de patología neoplásica
Huerta Aragonés J. Oncología para el pediatra de Atención Primaria (I): signos y síntomas sugerentes de patología neoplásica. Form Act Pediatr Aten Prim. 2014;7;4-15